bwa专题

IGV、BWA、Samtool的可视化分析过程。
1.首先使用BWA进行reads比对,结果会产生sam文件。 2.对结果文件进行可视化的时候必须要注意,IGV并不是支持所有的文件格式,所以要通过samtool进行格式转换。 3.所以比对后的sam文件不能够进行直接可视化,首先通过samtool软件进行格式转化为bam文件, samtools view -h file.bam > file.samsamtools view -b -S

生信学习笔记:用conda安装bwa、samtools和tophat2以及问题解决
用conda安装bwa、samtools和tophat2 bwa $ conda install bwa samtools $ conda install samtools tophat2 安装 wget http://ccb.jhu.edu/software/tophat/downloads/tophat-2.1.0.Linux_x86_64.tar.gz 解压 tar -zxvf

【一起学生信】 bwa -M 参数解读
bwa mem 比对时,会有一个 -M 参数,bwa官方给出的解释是 mark shorter split hits as secondary。 -M 参数用来处理同一个reads比对到参考基因组上不同位置的情况。 不加 -M 目前的比对,都不加 -M 如果不加入 -M 参数,这种情况bam中的 flag= 2048 ( supplementary alignment ) # 必须做
BWA BOEA HANA
BWA & BOEA & HANA 一、 BWA- Increases BW query ,supports only SAP Data. BWA accelerates data sourced from BWA which maybe from have been loaded from any data source. BWA 7.2 with BW 7.3
bwa、bowtie2、tophat、hisat2 比对软件学习中的笔记整理
对常用的比对软件学习进行用法整理记录。记录的内容相对简单,详细说明及用法还得参考软件使用说明书 bwa、bowtie2、tophat、hisatbwabwa(Burrows-Wheeler Aligner)bwa文档说明http://bio-bwa.sourceforge.net/bwa.shtmlBWA用于将低差异的序列映射到一个大的参考基因组,如人类基因组。由BWA-backtrack
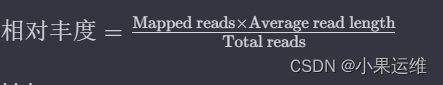
基于BWA,Bowtie2,samtools、checkm等工具计算宏基因组学序列分析中Contigs与Genes在样品中的丰度,多种计算方式和脚本对比
计算contigs和genes相对丰度可以提供有关微生物群落结构和功能的信息。以下是计算这两个指标的意义: 1. Contigs的相对丰度:contigs是利用基因组测序技术获得的碎片序列,通过计算contigs的相对丰度可以了解微生物群落中不同菌种的相对丰度。这可以帮助研究者理解微生物群落的物种组成和群落结构。 2. Genes的相对丰度:基因是生物体内功能的基本单位,通过计算基因的相对丰度

【bioinfo】bwa mem 比对分值参数测试
常用的序列比对软件bwa: command对应的多种命令,这里使用的是mem,即使用BWA-MEM算法进行序列比对。 bwa mem命令比对: 下方官网上介绍的mem命令: bwa mem 比对分值参数: 参数默认比对情况分值说明-A[1]Match11bp比对得1分-B[4]Mismatch-41bp错配扣4分-O[6,6]gap(ins,del)-6,-61bp的ins扣6分,d

BWA-MEM算法结构分析
一、BWA-MEM函数框架 软件位置:https://sourceforge.net/projects/bio-bwa/ 1 读入 bwt、options、reads; 2 利用mem_chain生成chain; 3 利用mem_chain_flt过滤掉部分chain; 4 利用mem_chain2aln生成比对结果数据。 1.第一步:数据输入 加载已经生成的bwt表。接口的参数

bwa计算wig文件
2019独角兽企业重金招聘Python工程师标准>>> bwa会输出mapping不上的reads例如: A00268:187:H5HVYDSXX:2:1101:22824:1532 77 * 0 0 * * 0 0 TGGGCCTCGGCCAGCGGGTTGATCAGCACCTGCTCGTT