本文主要是介绍【bioinfo】bwa mem 比对分值参数测试,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
-
常用的序列比对软件bwa:
command对应的多种命令,这里使用的是mem,即使用BWA-MEM算法进行序列比对。 -
bwa mem命令比对:
下方官网上介绍的mem命令:

-
bwa mem比对分值参数:

参数 默认 比对情况 分值 说明 -A [1] Match 1 1bp比对得1分 -B [4] Mismatch -4 1bp错配扣4分 -O [6,6] gap(ins,del) -6,-6 1bp的ins扣6分,del扣6分 -E [1] gap extension 1 发生extension罚分系数 -L [5,5] soft clipping -5,-5 在5’端,3’端的softclip扣5分 -U [17] unpaired read -17 不成对的read扣17分 -E: g+[1]*s (g:gap罚分值, s:gap的长度(从第1bp的gap开始算), [1]:该参数设定)-L: 按错配/indel,还是按softclip。 是选择分值高的作为最优比对。该分值设定越大,即对softclip的惩罚越大,那么选择最优比对时,更倾向于错配/indel(尽可能的比对上,而不是直接给出softclip),同样带来的弊端就是有很多错配。但是softclip的分值则不在AS中显示
-
测试分值相关参数:
Test1:
$ BWA mem -M -k10 -T10 ref.fa tst1.fq > tst1.sam $ cat ref.fa >seq1 CACGATGTCTCTCCTCTTAATGTGCTGCACATCTGTAGGATGGGGACAAA该测试主要针对序列的长度,以及有错配/Ins/Del的情况。

其中:
-M: mark shorter split hits as secondary (标记secodary比对结果);
-k: minimum seed length [19] (比对的序列最短长度,默认是19bp);这里设定10。这里的seed不是太明白,是否是指read序列,还是有别的含义?测试是序列可比对长度低于该阈值,则不显示比对结果。
-T: 比对的最小分值阈值,默认值30。即比对得分值<30不输出比对结果(结果显示未比对上),比对得分值>=30输出比对结果。这里设定10。[-k]参数的作用优于[-T],就是说,如果[-k]设定长度阈值较高(eg:19bp),而[-T]参数设置较低(eg:10,即分值最低为10),那么当read长度达不到19bp (但比对分值>=10),结果仍认为是未比对上。
ref.fa: fasta格式的参考序列文件;
tst1.fq: 输入的fq文件;
tst1.sam:输出sam格式比对结果文件。该测试其他分值参数使用的是默认值,以read id 为
2del2ins的序列为例计算分值:- CIGAR值得为:13M2D8M2I12M10S;(M:match;D:del;I:ins;S:softclip) - AS tag给出比对分值:"AS:i:17"(17分) - 分值计算:- match得分:13+8+12=33- indel罚分:(6+1*2)+(6+1*2) = 16 (ins和del各有两个)- 最终得分:33-16=17注意:这里的分值并没有计算softclip,因为softclip的分值是在比对过程中,用于计算:
是将一端的序列视为softclip还是视为可比对(有match、mistmach、indel);
如果按比对的分值>视为softclip分值,则选择比对上结果,反之,视为softclip。Test2:
测试靠近read两端位置出现错配, 比对结果是match/mismatch还是softclip?$ BWA mem -M ref.fa tst.fq > tst.sam $ cat ref.fa >ref TCGTCAATTCCAGGCCAGCCCATGAAGTGAGCAGA
比对结果中,分值计算:readID: "match"是完全match,碱基数35,分值是: 35; readID: "mis1"第三个碱基有一个碱基错配,比对上的碱基有34,得分值:34+(-4)=30;(奇怪:这里为什么给到分值(AS)是32?) readID:"mis1_ins1"是有一个碱基错配,和一个ins。- 如果按错配+ins,可match碱基是34个,得分是:34+(-4)+(-6)=24;- 如果从ins处及左端(5')视为6bp的softclip(罚5分),分值是:34+(-5)=29。- 也就是按softclip分值更高,所以比较结果给出的是softclip。
注意:bwa比对时,如果序列长度和碱基质量值不同,bwa比对结果中不显示该序列,也没有报错。
2021-06-08
对bwa比对的一些测试中,发现如果参考序列之间有相似时,要输出所有比对结果的话,情况可能不理想。
比如这是参考序列ref.fa:(随意编造的)
>ref1
CTACCGATCTGCACTGTAAGCACCCTTATG
>ref2
CTACCGATCTGCACTATAAGCACCCTTATA
>ref3
CTACCGATCTGCACGAACAGCACCCTTATG
>ref4
CTACCGATCTGCACTGTAAGCACCCTTATT
>ref5
CTACCGATCTGCACTATGAGCACCCTTATG
然后使用同样的序列作为test.fq,就是:
@ref1
CTACCGATCTGCACTGTAAGCACCCTTATG
+
FFFFFFFFFFFFFFFFFFFFFFFFFFFFFF
@ref2
CTACCGATCTGCACTATAAGCACCCTTATA
+
FFFFFFFFFFFFFFFFFFFFFFFFFFFFFF
@ref3
CTACCGATCTGCACGAACAGCACCCTTATG
+
FFFFFFFFFFFFFFFFFFFFFFFFFFFFFF
@ref4
CTACCGATCTGCACTGTAAGCACCCTTATT
+
FFFFFFFFFFFFFFFFFFFFFFFFFFFFFF
@ref5
CTACCGATCTGCACTATGAGCACCCTTATG
+
FFFFFFFFFFFFFFFFFFFFFFFFFFFFFF
进行比对:
bwa index -a is ref.fa # 构建索引
samtools faidx ref.fa # 构建索引
bwa mem -k1 -T1 ref.fa test.fq > test.sam # 默认输出结果
bwa mem -k1 -T1 -a ref.fa tmp.fq > test.all.sam # 比对输出所有结果
默认输出的结果:(候选比对结果会在XAtag中)

使用-a参数输出的结果:(与默认在XA中的相同)
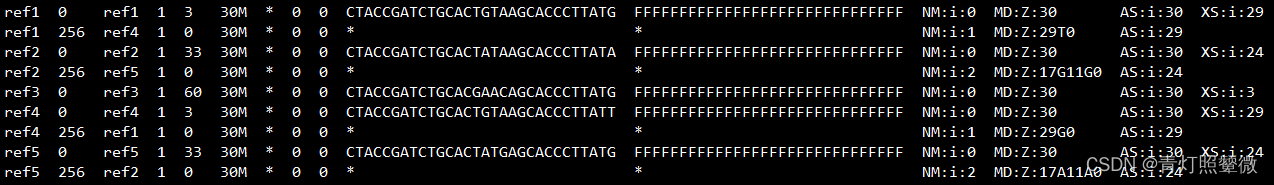
但是直观看的话,比如,ref1输出的比对只有ref1,ref4,直观看ref5也可以(2mis)

==但是,==构建ref时,只使用一个序列,比如只用ref5:
>ref5
CTACCGATCTGCACTATGAGCACCCTTATG
test.fq的文件比对结果则会是:

2023.2.23
这篇关于【bioinfo】bwa mem 比对分值参数测试的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!



