bam专题

全外显子测序分析流程3 - Exon.Interval.bed文件生成和BAM文件标记重复
全外显子测序分析流程3 - Exon.Interval.bed文件生成和BAM文件标记重复 分析流程步骤其他相关文章: Python处理生信分析流程配置文件4种方法 全外显子测序分析流程1 - Fastq质控与去接头、低质量和引物序列 全外显子测序分析流程2 - BWA-MEM比对到参考基因组与BAM统计 1. 封装流程特点 python封装, 参数控制配置文件设置核心参数,便于全流程

生信技能57 - Samtools获取指定外显子区域depth和提取BAM文件序列
1. Samtools depth 根据指定bed文件,获取指定区域的覆盖度信息。 # 提取IDT xGen V1 HBA1 exon bedcat xgen-exome-hyb-panel-v1-targets-hg19.bed|grep -w HBA1 > hba.exon.bed# 提取HBA1 外显子的覆盖度# -b: 提取depth的bed文件samtools depth -b

如何从BAM文件中提取fastq
虽然高通量测序分析最常用的操作是将fastq比对到参考基因组得到BAM文件,但偶尔我们也需要提取BAM文件中特定区域中fastq。最开始我认为这是一个非常简单的操作,因为samtools其实已经提供了相应的工具samtools fastq. 以biostar handbook的Ebola病毒数据为例,首先获取比对得到的BAM文件。 # 建立文件夹mkdir -p refs# 根据Acces
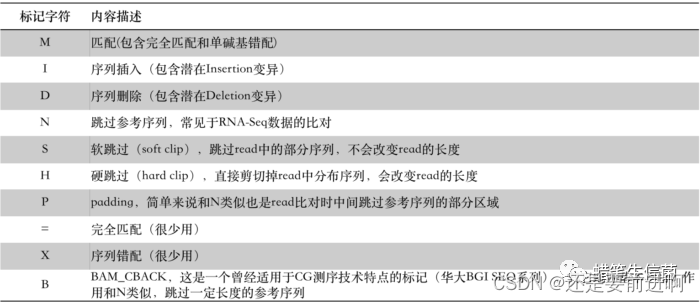
资料总结分享:SAM,bam,bed文件格式
目录 sam文件 bam文件 bed 文件 sam文件 SAM(Sequence Alignment/Map)文件是存储测序数据比对结果的一种常见格式。SAM文件通常用于存储DNA或RNA测序数据在参考基因组上的比对结果。 SAM文件由多行文本组成,每一行代表一个比对结果。SAM文件中的每一行包含了比对的序列ID、比对的标志、参考序列的名称、序列的起始位置、比对得分、序列的序

神经网络算法详解:反馈神经网络(Hopfield网络、双向联想记忆网络BAM、玻尔兹曼机BM、RBM)
本文介绍了反馈神经网络,包括Hopfield网络,离散Hopfield网络(DHNN),连续Hopfield网络(CHNN),双向联想记忆网络(BAM),玻尔兹曼机(BM),受限玻尔兹曼机(RBM)。其中对于BAM、BM、RBM只是对其进行了简单的介绍,并没有详细地推导算法。本文的目的旨在了解这些算法,先知道这些网络的改进和应用场景,当有业务需求的时候,再详细研究。 系列文章: 【神经网络

生物信息数据格式:sam,bam格式
文章目录 数据获取格式说明实例演练统计共有多少条reads(pair-end-reads这里算一条)参与了比对参考基因组统计共有多少种比对的类型(即第二列数值有多少种)及其分布筛选出比对失败的reads,看序列特征比对失败的reads区别成单端失败和双端失败情况,并且拿到序列ID筛选出比对质量值大于30的情况(看第五列)筛选出比对成功,但是并不是完全匹配的序列筛选出insert size长度
YoloV5改进策略:BAM瓶颈注意力模块|BAM详解以及代码注释|CBAM姊妹篇|有效涨点
论文:《BAM:瓶颈注意力模块》 https://arxiv.org/pdf/1807.06514.pdf 近期深度神经网络的进展主要通过架构搜索来增强其表示能力。在这项工作中,我们专注于注意力在一般深度神经网络中的作用。我们提出了一种简单而有效的注意力模块,名为瓶颈注意力模块(BAM),可以与任何前馈卷积神经网络集成。我们的模块沿两个独立的通道和空间路径推断注意力图。我们将模块放置在模型中的每
YoloV8改进策略:BAM瓶颈注意力模块|BAM详解以及代码注释|CBAM姊妹篇|有效涨点
论文:《BAM:瓶颈注意力模块》 https://arxiv.org/pdf/1807.06514.pdf 近期深度神经网络的进展主要通过架构搜索来增强其表示能力。在这项工作中,我们专注于注意力在一般深度神经网络中的作用。我们提出了一种简单而有效的注意力模块,名为瓶颈注意力模块(BAM),可以与任何前馈卷积神经网络集成。我们的模块沿两个独立的通道和空间路径推断注意力图。我们将模块放置在模型中的每

RNA-seq流程学习笔记(9)-使用RSeQC软件对生成的BAM文件进行质控
参考文章: 用RSeQC对比对后的转录组数据进行质控 高通量测序质控及可视化工具包RSeQC RSeQC使用笔记 1. 质控的原因及相关软件 在A survey of best practices for RNA-seq data analysis里面,提到了人类基因组应该有70%~90%的比对率,并且多比对read(multi-mapping reads)数量要少。另外比对在外显子和所比对链

samtools及bam文件的相关知识
文章目录 一些重要的概念首先是关于模板和read的概念然后是线性比对(linear alignment)和嵌合比对(chimeric alignment)的概念Phred scale1-base和0-base文件 bam文件的headerbam文件的主要内容 本文主要记录了阅读http://samtools.github.io/hts-specs/SAMv1.pdf 时学习到的一些内