本文主要是介绍R语言绘制动态网络图Network教程WGCNA,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
今天分享的笔记是使用NetworkD3对WGCNA的共表达网络进行可视化,创建交互式动态网络图,展示基因之间的相互关系,可以用于转录组或者其他调控网络展示。
加权基因共表达网络分析 (WGCNA, Weighted correlation network analysis)是用来描述不同样品之间基因关联模式的系统生物学方法,可以用来鉴定高度协同变化的基因集,并根据基因集的内连性和基因集与表型之间的关联鉴定候补生物标记基因或治疗靶点。
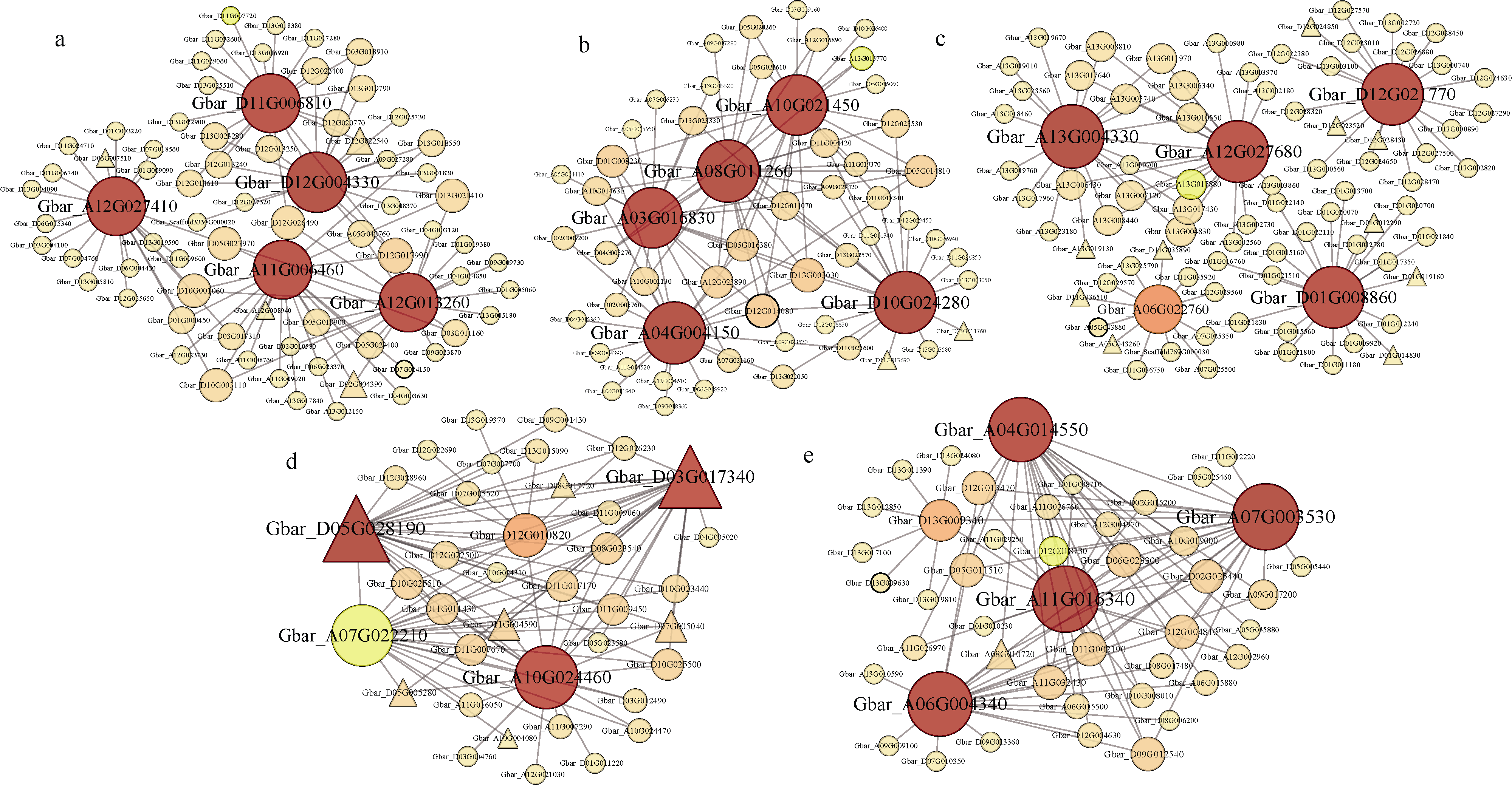
例如上图展示特异性模块内的基因共表达网络,点的大小和深浅代表该基因在网络中连通性的高低。转录因子用三角形表示, 其他基因用圆形表示。
如何绘制动态网络图?
首先,加载R包和数据,所用到的数据是WGCNA分析得到的输出网络文件,格式是Cytoscape的输入格式,本文的示例数据已上传,留言区回复邮箱,系统自动发送示例数据和全部代码。
library(networkD3)
library(tidyverse)
library(vroom)
df_node <- vroom::vroom("CytoscapeInput-nodes-black.txt")
df_edge <- vroom::vroom("CytoscapeInput-edges-black.txt")
> head(df_node)
# A tibble: 6 × 3
nodeName altName `nodeAttr[nodesPresent, ]`
<chr> <chr> <chr>
1 AT1G01010 AT1G01010 black
2 AT1G01090 AT1G01090 black
3 AT1G01180 AT1G01180 black
> head(df_edge)
# A tibble: 6 × 6
fromNode toNode weight direction fromAltName toAltName
<chr> <chr> <dbl> <chr> <chr> <chr>
1 AT1G69920 AT1G71030 0.445 undirected AT1G69920 AT1G71030
2 AT1G15125 AT1G71030 0.440 undirected AT1G15125 AT1G71030
3 AT1G02920 AT1G71030 0.438 undirected AT1G02920 AT1G71030
df_node文件保存节点信息,df_edge保存边的信息,包括起始位置和结束为止,以及连线的权重大小。这里每个节点可以表示一个基因,节点之间的weight权重值用来表示两个基因之间的关联性。
数据的过滤与筛选
df_edge <- df_edge %>% arrange(-weight) %>% head(100)
# 删除自身和自身相关位点
df_edge <- df_edge[which(df_edge$fromNode != df_edge$toNode),]
networkData <- df_edge[1:2]
simpleNetwork(networkData,linkDistance = 100)
由于基因数量比较多,因此这里先按照权重值进行排序,然后选取前100行,这一步可以根据你的需要设置,也可以按照制定阈值过滤,然后绘制一张简单版本的网络图:
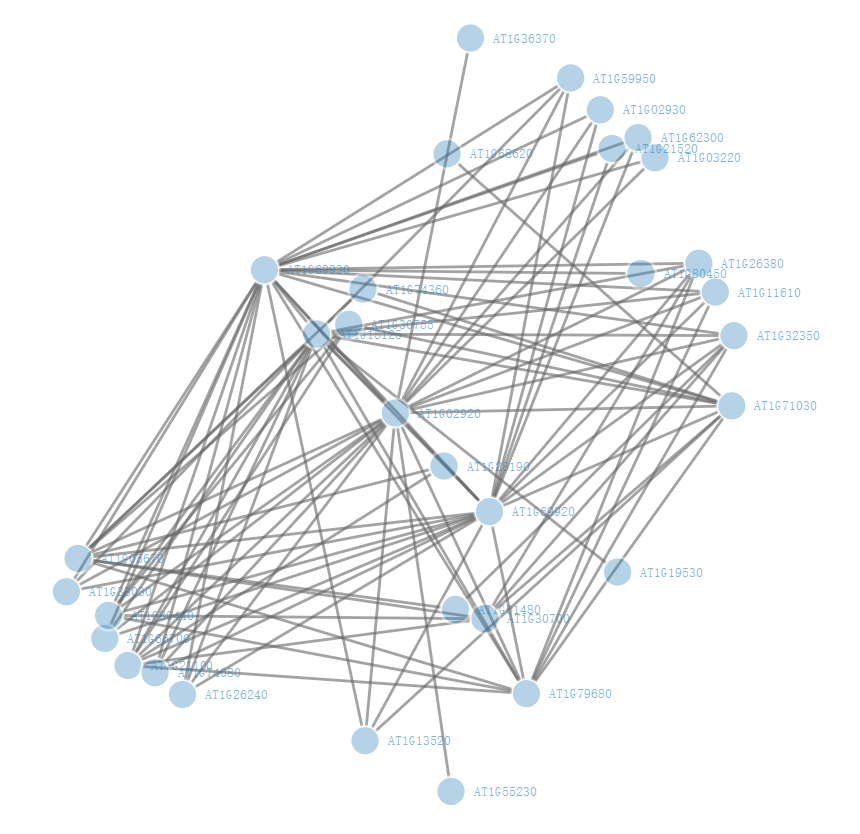
格式转换与重新编码
由于我们WGCNA输出的文件中节点都是通过基因ID来表示,但是绘图时无法直接识别节点ID,需要修改为数字0、1、2...因此,需要对节点进行重新修改。
# 转换格式
df_edge_net <- df_edge[,c(1,2,3)] %>% as.data.frame()
df_node_net <- df_node[,c(1,3)] %>% as.data.frame()
colnames(df_edge_net) <- c("source" ,"target" ,"value")
colnames(df_node_net) <- c("name","group")
# 合并第一列和第二列,并取并集
merged_elements <- union_all(df_edge_net$source,df_edge_net$target) %>% unique()
# 对合并后的元素进行编号
element_numbers <- seq_along(merged_elements)
# 创建一个新的数据框,包含合并的元素和对应的编号
result_df <- data.frame(merged_elements, element_numbers)
result_df$element_numbers <- result_df$element_numbers-1
# 使用映射表更新原始数据框的第一列和第二列
df_edge_net$source <- result_df$element_numbers[match(df_edge_net$source, result_df$merged_elements)]
df_edge_net$target <- result_df$element_numbers[match(df_edge_net$target, result_df$merged_elements)]
经过这一步处理后能够得到两个新的数据框,这就是绘制动态网络图的关键输入数据。在此基础上,我们还可以添加一些额外的信息,比如按照不同的分组将节点赋予不同的颜色,或者根据根据基因之间的正调控和负调控设置连接线的颜色。
# 生成模拟数据
df_edge_net$value <- c(runif(nrow(df_edge_net)/2,0,1),runif(nrow(df_edge_net)/2,0,5))
df_edge_net$color <- c(rep("red",50),rep("green",50))
value值表示节点之间连线的权重大小,可以用来展示两个基因之间的关联程度,该值越大线越粗,关联性越强。
color值可以用来设置连线的颜色,比如设置正调控为红色,负调控为绿色。
除了设置节点与节点之间边的关系,还能设置单个节点的参数,比如通过下面的代码设置节点的大小用来表示基因的表达量,表达量高的基因节点直径越大。还可以用过Type将节点进行分组,比如转录因子为A组,目标基因为B组等等。
df_node_net <- result_df
df_node_net$size <- runif(nrow(df_node_net),0,20)
df_node_net$type <- rep(c("A","B","C"),10000)[1:nrow(df_node_net)]
colnames(df_node_net) <- c("name", "group", "size","type")
绘制动态网络图
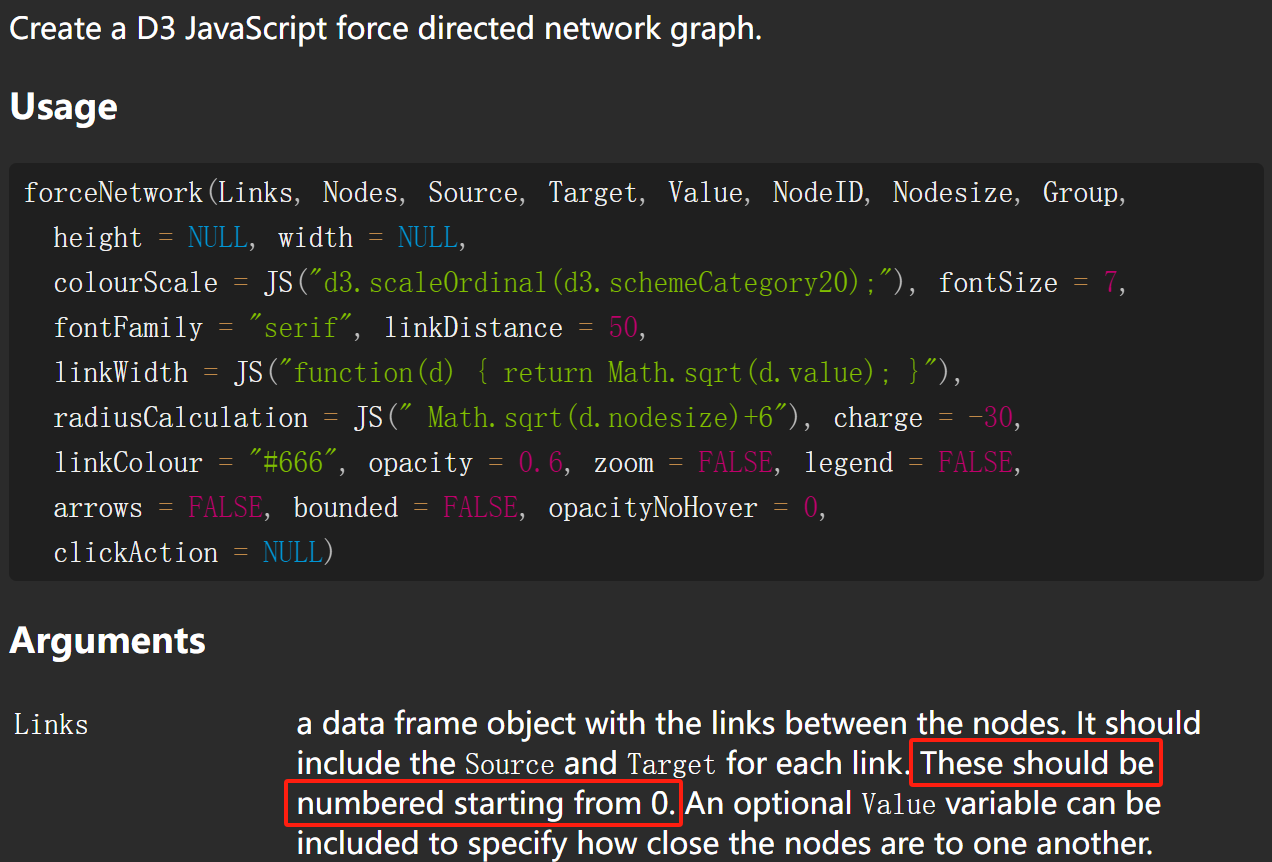
接下来通过调用forceNetwork绘制网络图,将刚刚的两个数据作为输入文件,设置如下参数即可获得结果图。
p <- forceNetwork(Links = df_edge_net,
Nodes = df_node_net,
Source = "source",
Target = "target",
linkColour=df_edge_net$color,
arrows=TRUE,
legend=TRUE,
Value = "value",
NodeID = "name",
Group = "type",
bounded=F,
opacityNoHover = 0.5,
linkDistance = 100,
charge=-500,
Nodesize='size',
# radiusCalculation = "Math.sqrt(d.nodesize,2)*5",
# linkWidth = JS("function(d) { return Math.sqrt(d.value)-4;}"),
# linkDistance=JS("function(d){return 1/(d.value)*100 }"),
opacity = 0.9,
zoom = T,
fontFamily = "Aril",
fontSize = 12)
p
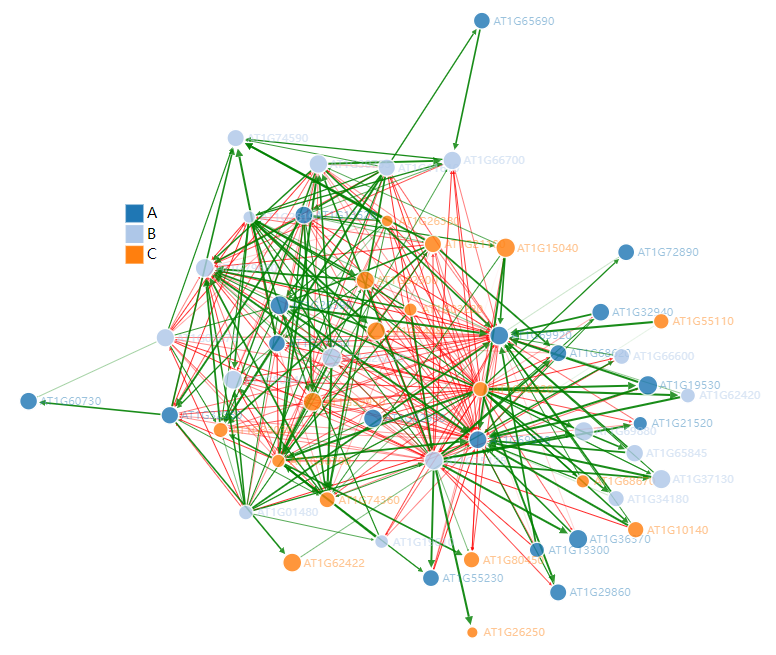
这张图是通过JS实现的,因此支持动态交互,比如将鼠标放在节点上会显示节点名称(基因ID),还可以拖动节点查看与之关联节点。
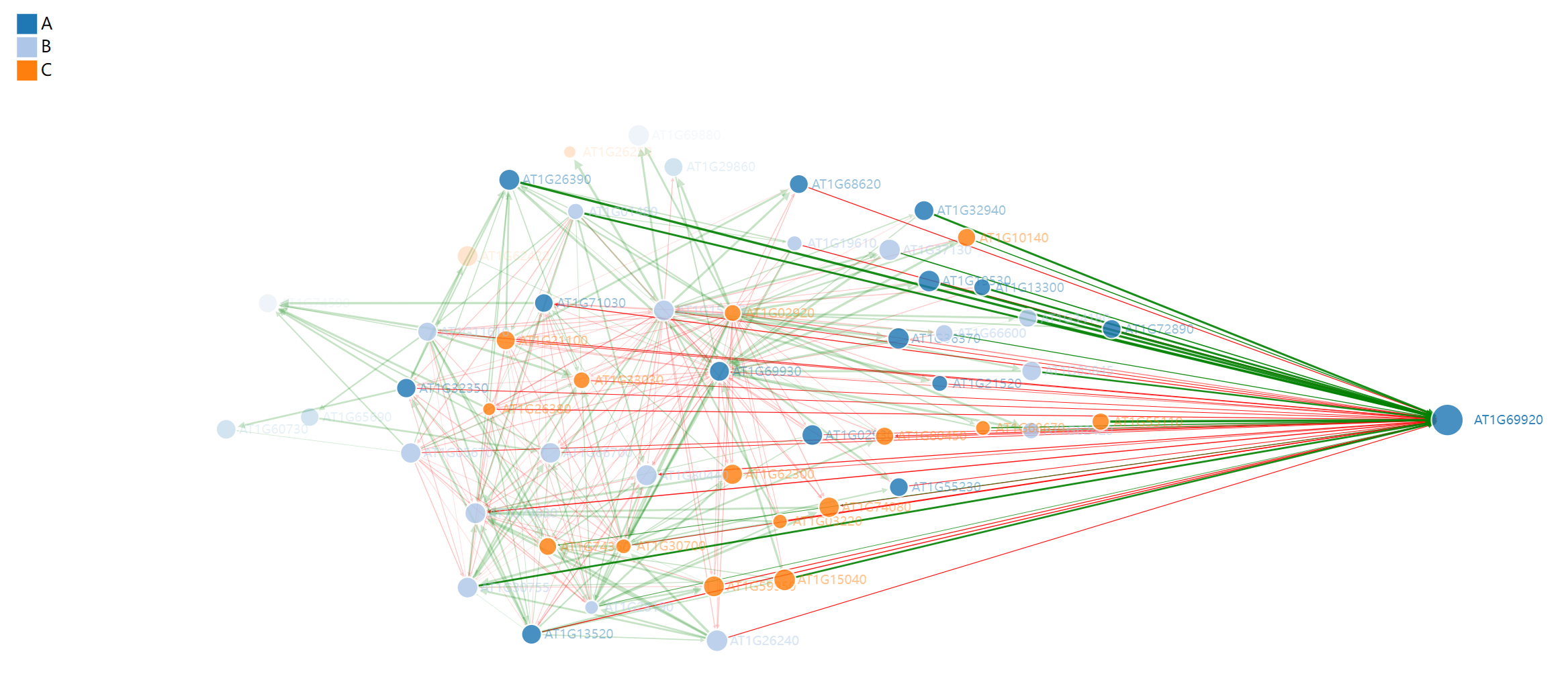
如果想要将其保存下来,最好的方法是html格式,这样仍具有动态交互属性。
saveNetwork(network = p,file = 'Net.html')
今天分享的内容就到这里,感谢您的阅读,如需本文代码和数据,请把收件邮箱发在评论区,欢迎点赞转发分享。
本文由 mdnice 多平台发布
这篇关于R语言绘制动态网络图Network教程WGCNA的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!







