本文主要是介绍资料总结分享:《全外显子测序数据的流程和原理》,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
1外显子与测序,生信流程
1.1 外显子是什么?
外显子是基因组中能够转录组出成熟RNA的部分。一个基因组中所有外显子的集合,即为外显子组。值得注意的是,通常所说的全外显子组测序,是指针对蛋白编码基因的外显子,很少涉及非编码基因。
外显子组(exome)是基因组中所有外显子的集合。人类拥有约18万个外显子,约占人类基因组的1%,即约3000万个bp(30MB)。
关于外显子,需要注意的一个特殊情况是非翻译区(UTR)。在mRNA的两侧分别存在5'UTR(前导序列)和3'UTR(尾部序列),它们的作用分别是调控翻译的启动和终止。它们由外显子序列构成,但不会被翻译成氨基酸。 所以,并非所有外显子序列都会被翻译成氨基酸。
1.2 全外显子组测序是什么?
全外显子组测序(Whole Exome Sequencing,WES)是一种高通量的DNA测序技术,用于分析一个个体的全外显子组,即所有外显子的序列。
外显子是基因组中编码蛋白质的区域,占据了相对较小的基因组部分,而大部分基因组序列是非编码DNA。WES技术通过选择性地寻找并测序这些外显子区域,可以快速而经济地获得一个个体的基因组信息。
1.3全外显子测序选择性寻找外显子的原理是什么?
全外显子测序选择性寻找外显子的原理基于外显子捕获(exome capture)技术,其主要原理是利用特异性的探针或引物将外显子区域从基因组中富集出来,从而实现对外显子的选择性测序。
以下是全外显子测序选择性寻找外显子的主要原理和步骤:
设计探针或引物: 首先,根据目标物种的基因组序列信息,设计一系列特异性的DNA探针或引物,这些探针或引物会针对外显子区域进行选择性的结合。
富集外显子: 探针或引物与外显子区域结合后,形成探针-外显子DNA复合物。通过不同的方法,如液相杂交(liquid-phase hybridization)或固相杂交(solid-phase hybridization),将这些复合物从样本中富集出来。这些方法通常会利用探针或引物与外显子DNA的互补配对来实现选择性富集。
去除非外显子DNA: 在富集外显子的过程中,非外显子区域的DNA大部分会被去除或减少,从而实现对外显子的选择性富集。
测序:最后,对富集后的外显子DNA进行高通量测序,通常采用Illumina等平台进行测序。由于外显子已经被富集,因此测序过程中主要测序的是外显子区域的DNA片段,从而获得外显子组的测序数据。
1.4全外显子组测序的工作流程是什么样的?
一个WES测序的工作流程,大体可以分为这3个部分:文库制备,测序,生信分析。文库制备通常包含这些步骤:样本处理,DNA提取,定量,建库,杂交捕获,扩增,质控。测序,目前的仪器包括国外Illumina公司测序平台,以及华大智造国产测序平台等。生信分析的流程通常包含这些步骤:质控,拼接比对,去重和重排,变异检测,降噪和过滤,注释等。常用的软件有FastQC,BWA,GATK,ANNOVAR等。一个完整的全外显子组测序,从样本处理到完成数据分析,通常需要10天左右时间。
1.5 生信分析流程
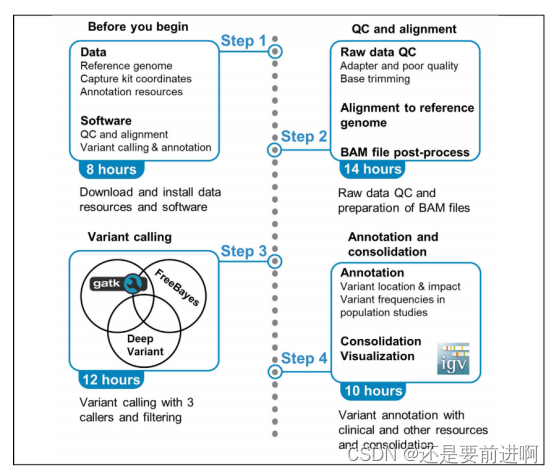
1、原始测序数据的质控。2、read比对,排序和去除重复序列。3、Indel区域重(“重新”的“重”)比对。4、碱基质量值重校正。5、变异检测。6、变异结果质控和过滤。
1.6 各步骤的原理
1.6.1.reads 比对
为什么需要比对?我们已经知道NGS测序下来的短序列(read)存储于FASTQ文件里面。虽然它们原本都来自于有序的基因组,但在经过DNA建库和测序之后,文件中不同read之间的前后顺序关系就已经全部丢失了。因此,FASTQ文件中紧挨着的两条read之间没有任何位置关系,它们都是随机来自于原本基因组中某个位置的短序列而已。
因此,我们需要先把这一大堆的短序列捋顺,一个个去跟该物种的参考基因组比较,找到每一条read在参考基因组上的位置,然后按顺序排列好,这个过程就称为测序数据的比对。这也是核心流程真正意义上的第一步,只有完成了这个序列比对我们才有下一步的数据分析。
1.6.2 排序
为什么需要排序,为什么BWA比对后输出的BAM文件是没顺序的!原因就是FASTQ文件里面这些被测序下来的read是随机分布于基因组上面的,第一步的比对是按照FASTQ文件的顺序把read逐一定位到参考基因组上之后,随即就输出了,它不会也不可能在这一步里面能够自动识别比对位置的先后位置重排比对结果。因此,比对后得到的结果文件中,每一条记录之间位置的先后顺序是乱的,我们后续去重复等步骤都需要在比对记录按照顺序从小到大排序下来才能进行,所以这才是需要进行排序的原因。
1.6.3 去重复
在排序完成之后我们就可以开始执行去除重复(准确来说是 去除PCR重复序列)的步骤了。首先,我们需要先理解什么是重复序列,它是如何产生的,以及为什么需要去除掉?要回答这几个问题,我们需要再次理解在建库和测序时到底发生了什么。我们在前面文章中已经知道,在NGS测序之前都需要先构建测序文库:通过物理(超声)打断或者化学试剂(酶切)切断原始的DNA序列,然后选择特定长度范围的序列去进行PCR扩增并上机测序。因此,这里重复序列的来源实际上就是由PCR过程中所引入的。因为所谓的PCR扩增就是把原来的一段DNA序列复制多次。可是为什么需要PCR扩增呢?如果没有扩增不就可以省略这一步了吗?情况确实如此,但是很多时候我们构建测序文库时能用的细胞量并不会非常充足,而且在打断的步骤中也会引起部分DNA的降解,这两点会使整体或者局部的DNA浓度过低,这时如果直接从这个溶液中取样去测序就很可能漏掉原本基因组上的一些DNA片段,导致测序不全。而PCR扩增的作用就是为了把这些微弱的DNA多复制几倍乃至几十倍,以便增大它们在溶液中分布的密度,使得能够在取样时被获取到。所以这里大家需要记住一个重点,PCR扩增原本的目的是为了增大微弱DNA序列片段的密度,但由于整个反应都在一个试管中进行,因此其他一些密度并不低的DNA片段也会被同步放大,那么这时在取样去上机测序的时候,这些DNA片段就很可能会被重复取到相同的几条去进行测序,但是由同一个模板分子扩增出来的重复子文库只对应单一模板,在分析过程中应将重复片段予以去除。具体可查询下建库原理和测序原理。
1.6.4 局部重比对
局部重比对的目的是将BWA比对过程中所发现有 潜在序列插入或者序列删除(insertion和deletion,简称Indel)的区域进行重新校正。这个过程往往还会把一些已知的Indel区域一并作为重比对的区域,但为什么需要进行这个校正呢?
其根本原因来自于参考基因组的序列特点和BWA这类比对算法本身,注意这里不是针对BWA,而是针对所有的这类比对算法,包括bowtie等。这类在全局搜索最优匹配的算法在存在Indel的区域及其附近的比对情况往往不是很准确,特别是当一些存在长Indel、重复性序列的区域或者存在长串单一碱基(比如,一长串的TTTT或者AAAAA等)的区域中更是如此。
另一个重要的原因是在这些比对算法中,对碱基错配和开gap的容忍度是不同的。具体体现在罚分矩阵的偏向上,例如,在read比对时,如果发现碱基错配和开gap都可以的话,它们会更偏向于错配。但是这种偏向错配的方式,有时候却还会反过来引起错误的开gap!这就会导致基因组上原本应该是一个长度比较大的Indel的地方,被错误地切割成多个错配和短indel的混合集,这必然会让我们检测到很多错误的变异。而且,这种情况还会随着所比对的read长度的增长(比如三代测序的Read,通常都有几十kbp)而变得越加严重。
因此,我们需要有一种算法来对这些区域进行局部的序列重比对。这个算法通常就是大名鼎鼎的Smith-Waterman算法,它非常适合于这类场景,可以极其有效地实现对全局比对结果的校正和调整,最大程度低地降低由全局比对算法的不足而带来的错误。而且GATK的局部重比对模块,除了应用这个算法之外,还会对这个区域中的read进行一次局部组装,把它们连接成为长度更大的序列,这样能够更进一步提高局部重比对的准确性。
1.6.5 碱基质量校正
变异检测是一个极度依赖测序碱基质量值的步骤。因为这个质量值是衡量我们测序出来的这个碱基到底有多正确的重要(甚至是唯一)指标。它来自于测序图像数据的base calling。因此,基本上是由测序仪和测序系统来决定的。但不幸的是,影响这个值准确性的系统性因素有很多,包括物理和化学等对测序反应的影响,甚至连仪器本身和周围环境都是其重要的影响因素。当把所有这些东西综合在一起之后,往往会发现计算出来的碱基质量值要么高于真实结果,要么低于真实结果。我们寻找的就是突变,如果碱基质量测得不高,不准,那突变假阳性不就太高了吗。
本文较为简单,属于科普入门级别,想要再次深入了解,可看专业的文章和书籍。
资料来源:
1.网址:https://mp.weixin.qq.com/s/35QIeXBV6myy4BpHFsj5nA,https://mp.weixin.qq.com/s/y42k6dnUevCTxctllpWx3g
2.文章: 《Protocol for unbiased, consolidated variant calling from whole exome sequencing data》
本文是我收集资料总结而来,资料来源已表明,之作学习交流使用,不为获利,如有侵权,联系立删。
这篇关于资料总结分享:《全外显子测序数据的流程和原理》的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!






