本文主要是介绍windows ubuntu子系统,单细胞篇 1.cellranger安装与分析,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
这几天,我将单细胞测序在windows ubuntu子系统中跑了一遍,将过程分享給大家。
单细胞测序
conda create -n 10xdb #创建环境
conda activate 10xdb
conda install -c bioconda cellranger -y #失败,可能源中没有
wget -O cellranger-7.2.0.tar.gz "https://cf.10xgenomics.com/releases/cell-exp/cellranger-7.2.0.tar.gz?Expires=1702235980&Key-Pair-Id=APKAI7S6A5RYOXBWRPDA&Signature=c5ZVKLpCggvKE5O1o0pR7X-IfMXAkWZdQ9o-BipsD9PQJP31eIR2vpy2J2Ucd5faQyM0ZgjsO98qdF616z62D4SRvJDHeYkdQzGSmoZNzcWRLM1Getvend4HkSxPxjePb5MhvohRKsh9~wR2RVivMSZXpiy6zhH1efwcF4-PPmSwJshbhU3pI7InTt5qg92kasL3ekv~8D-scp3kAiFwm15d-ufvblrox8g4P9JQjFHHDjLuu0F~P8-Qc31lMdOe~5KoDCbkE~UI7sEUBJktD0MR~lZ620OcYKgvaqCB-wLgI3VI0rH8RPxYvbgUXryBHnB0wEmqjaessFlNvhql1Q__"#下载成功
tar -xzvf cellranger-7.2.0.tar.gz #tar 解压缩
vim ~/.bashrc
export PATH="/mnt/h/softwore/cellranger-7.2.0/bin/:$PATH" ,source ~/.bashrc,#成功
#下载人源比对基因组
wget "https://cf.10xgenomics.com/supp/cell-exp/refdata-gex-GRCh38-2020-A.tar.gz" #这里下载太慢
在windows环境中下载
md5sum refdata-gex-GRCh38-2020-A.tar.gz
tar -xzvf refdata-gex-GRCh38-2020-A.tar.gz
mkdir 1.sra 2.raw_fastq 3.cellranger,cd 1.sra,touch download_file, #创建目录,创建文件。
vim download_file #编辑文件,文件名。
SRR7722941
SRR7722942
cat download_file |while read id;do (prefetch $id &);done #NCBI下载单细胞数据。下载方法和软件可见,最好早上下载。
3.windows下Ubuntu,sratoolkit软件,从ncbi的sra数据库下载数据。_sratools下载数据-CSDN博客
下载完数据后,将SRR数据转为fastq格式。
ls SRR* | while read id;do ( nohup fasterq-dump -O ./ --split-files -e 6 ./$id --include-technical & );done
#使用"ls SRR*"查找以"SRR"开头的文件,然后使用"while read id"逐行读取这些文件名,对每个文件执行以下操作:使用fasterq-dump命令下载SRA文件,将其转换成FASTQ格式,并将结果保存在当前目录下。使用--split-files选项将每个样本的双端序列拆分成两个文件。使用-e 6选项指定最大重试次数为6。使用--include-technical选项包括技术序列。使用&符号将每个命令放入后台执行,以便同时处理多个文件,加快处理速度。
#对10X的fq文件运行CellRangerd的counts流程,我们只做一个文件,首先,我们先改名。
mv SRR7722937_1.fastq.gz SRR7722937_S1_L001_I1_001.fastq.gz,
mv SRR7722937_2.fastq.gz SRR7722937_S1_L001_R1_001.fastq.gz,
mv SRR7722937_3.fastq.gz SRR7722937_S1_L001_R2_001.fastq.gZ
#每个SRR分出三个fastq文件,查阅资料是
- SRR7722941_S1_L001_I1_001.fastq:这是Index序列文件,包含了每个序列的索引信息。
- SRR7722941_S1_L001_R1_001.fastq:这是Read 1序列文件,包含了每个序列的第一部分。
- SRR7722941_S1_L001_R2_001.fastq:这是Read 2序列文件,包含了每个序列的第二部分。
#接下来运行Cellranger count流程
ref=/mnt/h/db/cellranger.db/refdata-gex-GRCh38-2020-A
id=SRR7722941
cellranger count --id=$id \
--transcriptome=$ref \
--fastqs=/mnt/h/sxfx/2023.1210.db/2.raw_fastq \
--sample=$id \
--nosecondary \
--localmem=15 \
--localcores=15
-
cellranger count:这是Cell Ranger软件的一个子命令,用于对单细胞RNA测序数据进行计数。
-
--id=$id:指定输出结果的唯一标识符,通常是分析的样本名称或编号,指定输出文件夹的名字。
-
--transcriptome=$ref:指定参考转录组的路径或者名称。这个参数告诉Cell Ranger在分析中使用哪个转录组参考序列。
-
--fastqs=/mnt/h/sxfx/2023.1210.db/2.raw_fastq:指定原始FASTQ文件的路径。这个参数告诉Cell Ranger去哪里查找原始的测序数据。
-
--sample=$id:指定样本名称或编号。这个参数用于告诉Cell Ranger如何标识分析中的每个样本。
-
--nosecondary:这个参数告诉Cell Ranger在对数据进行比对和计数时不考虑次要的比对结果。次要比对结果是指在一个位置有多个可能的比对结果时,除了最佳的比对结果外的其他比对结果。
-
--localmem=15:指定本地内存的使用量,单位为GB。这个参数告诉Cell Ranger在运行过程中最多可以使用多少内存。
-
--localcores=15:指定本地CPU核心数。这个参数告诉Cell Ranger在运行过程中最多可以使用多少CPU核心。
#最终结果在outs中
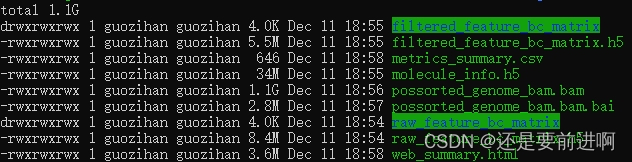
结果解读(参考生信技能树Jimmy的帖子):
-
web_summary.html:必看,官方说明 summary HTML file ,包括许多QC指标,预估细胞数,比对率等;
-
metrics_summary.csv:CSV格式数据摘要,可以不看;
-
possorted_genome_bam.bam:比对文件,用于可视化比对的reads和重新创建
FASTQ文件,可以不看; -
possorted_genome_bam.bam.bai:索引文件;
-
filtered_gene_bc_matrices:是重要的一个目录,下面又包含了 barcodes.tsv.gz、features.tsv.gz、matrix.mtx.gz,是下游Seurat、Scater、Monocle等分析的输入文件,是经过
Cell Ranger过滤后构建矩阵所需要的所有文件; -
filtered_feature_bc_matrix.h5:过滤掉的barcode信息HDF5 format,可以不看;
-
raw_feature_bc_matrix:原始barcode信息,未过滤的可以用于构建矩阵的文件,可以不看;
-
raw_feature_bc_matrix.h5:原始barcode信息HDF5 format,可以不看;
-
analysis:数据分析目录,下面又包含聚类clustering(有graph-based & k-means)、差异分析diffexp、主成分线性降维分析pca、非线性降维tsne,因为我们自己会走Seurat流程,所以不用看;
-
molecule_info.h5:可用于整合多样本,使用
cellranger aggr函数; -
cloupe.cloupe:官方可视化工具Loupe Cell Browser 输入文件,无代码分析的情况下使用,会代码的同学通常用不到。
这是我最近跑的一个流程,说实在的,博大精深,以后我会看一些文献,分享一下,流程好跑,背景知识很难啊。
这篇关于windows ubuntu子系统,单细胞篇 1.cellranger安装与分析的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!





