本文主要是介绍GSVA计算基因样本的相似性,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
下载包
# source('http://bioconductor.org/biocLite.R')
# options(BioC_mirror='http://mirrors.ustc.edu.cn/bioc/')
# biocLite('estimate')
# library(GSVA)
# browseVignettes('GSVA')
# browseVignettes('estimate')source("http://www.bioconductor.org/biocLite.R")
options(BioC_mirror="http://mirrors.ustc.edu.cn/bioc/",repos=structure(c(CRAN="http://mirrors.tuna.tsinghua.edu.cn/CRAN/")))
# biocLite('GSVA')
# biocLite('GSVAdata')
# biocLite('genefilter')
# biocLite('limma')
# biocLite('edgeR')#browseVignettes('GSVA') #get the GSVA code
# browseVignettes('estimate')
加载调用包
library(BiocGenerics)
library(parallel)
library(Biobase)
library(annotate)
library(AnnotationDbi)
library(stats4)
library(IRanges)
library(S4Vectors)
library(XML)
library(graph)
library(GSEABase)library(Biobase)
library(genefilter)
library(limma)
library(RColorBrewer)
library(org.Hs.eg.db)
library(hgu95a.db)
library(GSVAdata)
library(GSVAdata)
library(GSVA)
表达式数据转换
# input=read.table("FULLDATA.csv", header=TRUE, row.names="id", sep = ",")
# esete <- as.matrix(input)
# group<-read.csv("group.csv", row.names="sampleID", sep=",")
# metaData <- data.frame(labelDescription=c("group"),row.names=c("group"))
# AnnotatedDataFrame()
# AnnotatedDataFrame(data=group)
# AnnotatedDataFrame(data=group, varMetadata=metaData)
# anno <- as(group, "AnnotatedDataFrame")
# ExpressionSet()
# exset <- ExpressionSet(esete,anno)
绘制图形
(代码略)图形类似
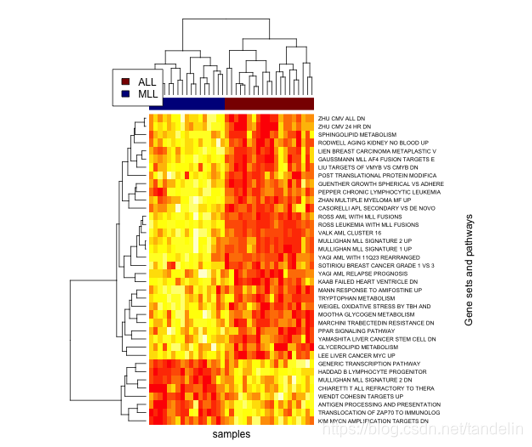
这篇关于GSVA计算基因样本的相似性的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!








