本文主要是介绍GenomeScope——jellyfish k-mer分析的下游分析,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
GenomeScope 2.0是一个可对多倍体基因组数据进行概括性分析的有效工具,其可以将jellyfish或KMC等k-mer分析软件的结果作为输入,并拟合混合模型,对基因组数据进行更深入的挖掘。
一、安装
GenomeScope 2.0的工作依赖于R,且需要安装其依赖包:argparse、minpack.lm。可以使用conda工具进行安装:
#安装依赖包
conda install -c conda-forge r-base r-minpack.lm r-argparse
安装完依赖包后需要将Github的相关文件克隆到工作目录下:
(有些时候会因为链接时间太久,导致克隆失败,可以尝试直接下载源代码)
#克隆工作环境
git clone https://github.com/tbenavi1/genomescope2.0.git
现在,便可以进入genomescope2.0目录,使用R脚本install.R安装GenomeScope 2.0了。
#进入工作目录
cd genomescope2.0
#运行R脚本安装GenomeScope2.0
Rscript install.R
二、使用
在使用之前需要认识以下几个重要的参数:
| 参数 | 功能 |
|---|---|
-i | 输入文件名 |
-o | 输出目录名 |
-k | k-mers的k值 |
-p | 基因组的倍性,默认为2 |
现在,便可以使用其主要脚本文件genomescope.R对.histo进行进一步分析了:
#进一步分析S_oblata_WGS_single.histo
genomescope2.0/genomescope.R -i S_oblata_WGS_single.histo -o GS_WGS -k 25
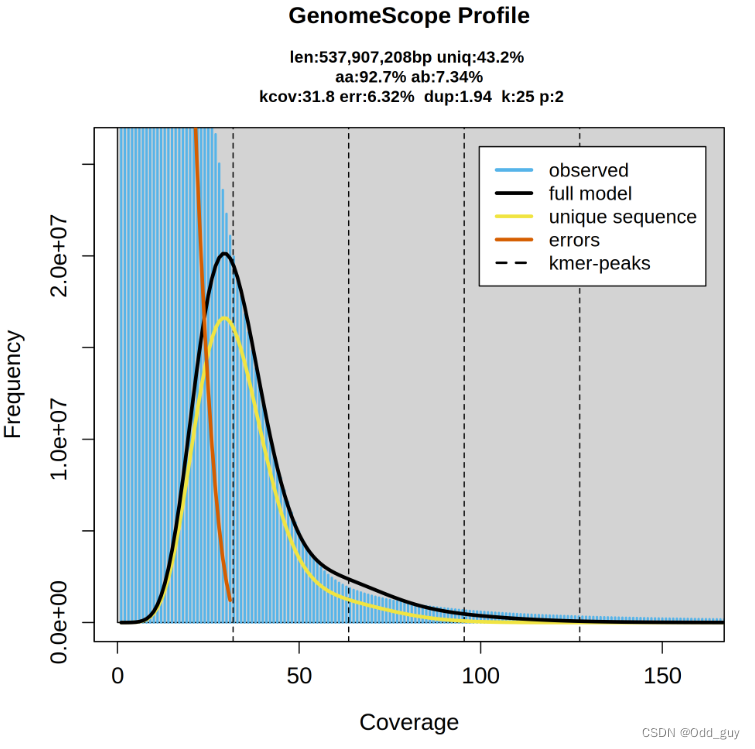
当出现:
GenomeScope analyzing S_oblata_WGS_single.histo p=2 k=25 outdir=GS_WGS
aa:92.7% ab:7.34%
Model converged het:0.0734 kcov:31.8 err:0.0632 model fit:1.94 len:537907208
即说明分析已经完成,其结果保存在GS_WGS目录下。
但是,由于文章中并没有提供具体的步骤,所以我得到的结果具有较高的错误率,可能是未进行数据过滤导致的。
Ending!!!
相关文章:
Ranallo-Benavidez, T.R., Jaron, K.S. & Schatz, M.C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nature Communications 11, 1432 (2020). https://doi.org/10.1038/s41467-020-14998-3
Ma, B., Wu, J., Shi, TL. et al. Lilac (Syringa oblata) genome provides insights into its evolution and molecular mechanism of petal color change. Commun Biol 5, 686 (2022). https://doi.org/10.1038/s42003-022-03646-9
这篇关于GenomeScope——jellyfish k-mer分析的下游分析的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!





