本文主要是介绍Paper reading (三十七):Generating focused molecule libraries for drug discovery with RNN,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
论文题目:Generating focused molecule libraries for drug discovery with recurrent neural networks
scholar 引用:203
页数:12
发表时间:2017.12
发表刊物:ASC(American Chemical Society) Central Science
作者:Marwin H. S. Segler, Thierry Kogej,Christian Tyrchan,§ and Mark P. Waller
摘要:
In de novo drug design, computational strategies are used to generate novel molecules with good affinity to the desired biological target. In this work, we show that recurrent neural networks can be trained as generative models for molecular structures, similar to statistical language models in natural language processing. We demonstrate that the properties of the generated molecules correlate very well with the properties of the molecules used to train the model. In order to enrich libraries with molecules active toward a given biological target, we propose to fine-tune the model with small sets of molecules, which are known to be active against that target. Against Staphylococcus aureus(金黄色葡萄球菌), this model reproduced 14% of 6051 holdout test molecules that medicinal chemists designed, whereas against Plasmodium falciparum(Malaria) 恶性疟原虫(疟疾), it reproduced 28% of 1240 test molecules. When coupled with a scoring function, our model can perform the complete de novo drug design cycle to generate large sets of novel molecules for drug discovery.
- 不太理解reproduce在这两种情况下的含义。
结论:
- we have shown that recurrent neural networks based on the long short-term memory (LSTM) can be applied to learn a statistical chemical language model.
- This can be used to generate libraries for virtual screening. 应用1
- we demonstrated that the model performs transfer learning when fine-tuned to smaller sets of molecules active toward a specific biological target, which enables the creation of novel molecules with the desired activity. 另一种应用
- we do not even need a set of known active molecules to start our procedure with
- three main advantages of our method:
- it is conceptually orthogonal to established molecule generation approaches
- our method is very simple to set up, to train, and to use
- it merges structure generation and optimization in one model.
- A weakness of our model is interpretability.
- extend our work: a small step to cast molecule generation as a reinforcement learning problem
- we believe that deep neural networks can be complementary to established approaches in drug discovery.
Introduction:
- One of the many challenges in drug design is the sheer size of the search space for novel molecules.
- Virtual screening is a commonly used strategy to search for promising molecules among millions of existing or billions of virtual molecules.
- in any molecular design task, the computer has to: create molecules, score and filter them, and search for better molecules, building on the knowledge gained in the previous steps.
- the generation of novel molecules: One strategy is to build molecules from predefined groups of atoms or fragments. another established approach is to conduct virtual chemical reactions based on expert coded rules
- we have recently shown that the predicted reactions from these rule-based expert systems can sometimes fail.
- scoring molecules and filtering out undesired structures: Target prediction classifies molecules into active and inactive, and quantitative structure–activity relationships (QSAR)
- the mapping from a target property value y to possible structures X is one-to-many
- In this work, we suggest a complementary, completely data-driven de novo drug design approach.
- we highlight the analogy of language and chemistry, and show that RNNs can also generate reasonable molecules.
- we demonstrate that RNNs can also transfer their learned knowledge from large molecule sets to directly produce novel molecules that are biologically active by retraining the models on small sets of already known actives.
- We test our models by reproducing hold-out test sets of known biologically active molecules.
正文组织架构:
1. Introduction
2. Methods
2.1 Representing Molecules
2.2 Language Models and Recurrent Neural Networks
2.3 Transfer Learning
2.4 Target Prediction
2.5 Data
2.6 Model Evaluation
3. Results and Discussion
3.1 Training the Recurrent Network
3.2 Generating Novel Molecules
3.3 Generating Active Drug Molecules and Focused Libraries
3.4 Simulating Design-Synthesis-Test Cycles
3.5 Why Does the Model Work?
4. Conclusion
正文部分内容摘录:
2. Methods
2.1 Representing Molecules
-
the SMILES format: a formal grammar which describes molecules with an alphabet of characters
-
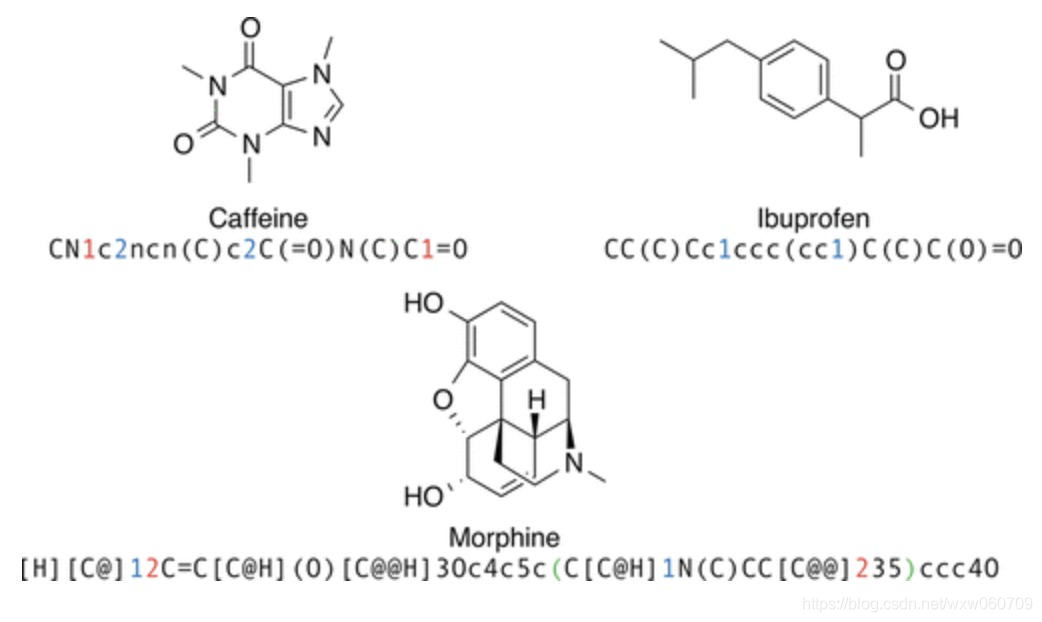
2.2 Language Models and Recurrent Neural Networks
- Language models are implemented,language models do not have to use words.
- characters in the SMILES alphabet, which form a (formal) language
- RNNs maintain state which is needed to keep track of the symbols seen earlier in the sequence.
- As the specific RNN function, in this work, we use the long short-term memory (LSTM)
- To encode the SMILES symbols as input vectors xt, we employ the “one-hot” representation.
- each molecule in our training data finishes with an “end of line” (EOL) symbol
- In this work, we used a network with three stacked LSTM layers, using the Keras library.
- The model was trained with back-propagation through time
- using the ADAM optimizer at standard settings.
- To mitigate the problem of exploding gradients during training, a gradient norm clipping of 5 is applied.
2.3 Transfer Learning
- To generate focused molecule libraries, we first train on a large, general set of molecules, then perform fine-tuning on a smaller set of specific molecules, and after that start the sampling procedure.
2.4 Target Prediction
-
To predict whether the generated molecules are active toward the biological target of interest, target prediction models (TPMs) were trained for all the tested targets
-
We evaluated random forest, logistic regression, (deep) neural networks, and gradient boosting trees (GBT) as models with ECFP4 (extended connectivity fingerprint with a diameter of 4) as the molecular descriptor. We found that GBTs slightly outperformed all other models and used these as our virtual assay in all studies GBT, 老朋友啊。。。
2.5 Data
- The chemical language model was trained on a SMILES file containing 1.4 million molecules from the ChEMBL database, which contains molecules and measured biological activity data.
2.6 Model Evaluation
- To evaluate the models for a test set T, and a set of molecules GN generated from the model by sampling, we report the ratio of reproduced molecules and enrichment over random (EOR)
- Intuitively, EOR indicates how much better the fine-tuned models work when compared to the general model.
3. Results and Discussion
In this work, we address two points: First, we want to generate large sets of diverse molecules for virtual screening campaigns. Second, we want to generate smaller, focused libraries enriched with possibly active molecules for a specific target.
这就像machine translation的通用翻译引擎和垂直领域翻译引擎。。。
3.1 Training the Recurrent Network
- a recurrent neural network with three stacked LSTM layers, each with 1024 dimensions, and each one followed by a dropout layer, with a dropout ratio of 0.2, to regularize the neural network.
- The model was trained until convergence, using a batch size of 128. The RNN was unrolled for 64 steps. It had 21.3 × 106 parameters.
- Within a few 1000 steps, the model starts to output valid molecules
3.2 Generating Novel Molecules
- To generate novel molecules, 50,000,000 SMILES symbols were sampled from the model symbol-by-symbol. This corresponded to 976,327 lines, from which 97.7% were valid molecules after parsing with the CDK toolkit.
- Removing all molecules already seen during training yielded 864,880 structures. After filtering out duplicates, we obtained 847,955 novel molecules.
- The created structures are not just formally valid but also mostly chemically reasonable.
- In order to check if the de novo compounds could be considered as valid starting points for a drug discovery program, we applied the internal AstraZeneca filters.
- Results: the algorithm generates preponderantly valid screening molecules and faithfully reproduces the distribution of the training data.
- To determine whether the properties of the generated molecules match the properties of the training data from ChEMBL, we followed the procedure of Kolb
- Results: Both sets overlap almost completely, which indicates that the generated molecules very well recreate the properties of the training molecules.
- Furthermore, we analyzed the Bemis–Murcko scaffolds of the training molecules and the sampled molecules.
- Results: indicates that the language model does not just modify side chain substituents but also introduces modifications at the molecular core.
3.3 Generating Active Drug Molecules and Focused Libraries
-
Targeting the
Receptor
-
Already after 4 epochs of fine-tuning the model produced a set in which 50% of the molecules are predicted to be active.
-
our method could deliver not only close analogues but also new chemotypes or scaffold ideas to a drug discovery project
-
To have the best of both worlds, that is, diverse and focused molecules, we therefore suggest to sample after each epoch of retraining and not just after the final epoch.
-
Targeting Plasmodium falciparum (Malaria)
-
To probe our model on this important target, we used a more challenging validation strategy.
-
this set was split randomly into a training (1239 molecules) and a test set (1240 molecules). The chemical language model was then fine-tuned on the training set. 7500 molecules were sampled after each of the 20 epochs of refitting.
-
This yielded 128,256 unique molecules. Interestingly, we found that our model was able to “redesign” 28% of the unseen molecules of the test set.
-
Targeting Staphylococcus aureus (Golden Staph)
-
our model could retrieve 14% of the 6051 test molecules.
3.4 Simulating Design-Synthesis-Test Cycles
- in combination with a target prediction or scoring model, our model can at least simulate the complete de novo design cycle.
3.5 Why Does the Model Work?
- sampling from this model does not help much if we want to generate actives for a specific target
- while the model often seems to have made small replacements in the underlying SMILES, in many cases it also made more complex modifications or even generated completely different SMILES.
- Many rediscovered molecules are in the medium similarity regime.
- A plausible explanation why the model works is therefore that it can transfer the modifications that are regularly applied when series of molecules are studied, to the molecules it has seen during fine-tuning.
这篇关于Paper reading (三十七):Generating focused molecule libraries for drug discovery with RNN的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!


![The `XXXUITests [Debug]` target overrides the `ALWAYS_EMBED_SWIFT_STANDARD_LIBRARIES` build......](https://i-blog.csdnimg.cn/blog_migrate/7e43f76c8b7367452a401d20f5dce54e.png)




