本文主要是介绍使用vsearch进行16s扩增子高通量序列分析步骤,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
1、vsearch分析工具介绍:
VSEARCH是一个开源免费的64位,无内存限制的扩增子数据处理分析软件。(点到为止,其他的建议大家参考原文献和网站)
github:GitHub - torognes/vsearch: Versatile open-source tool for microbiome analysis
最新文献:Edgar RC (2016) UNOISE2: improved error-correction for Illumina 16S and ITS amplicon sequencing. bioRxiv. doi:10.1101/081257
二进制文件下载(直接复制到目录就可以开始运行的):Release VSEARCH 2.23.0 · torognes/vsearch · GitHub
2、vsearch 安装:
建议大家直接下载二进制文件,github有时候不通,可以使用本站链接下载
https://download.csdn.net/download/zrc_xiaoguo/88404546
注意事项:无论使用编译安装还是使用二进制直接复制运行,都要注意安装对应版本的依赖库,出现报错时参考一下github里安装指定依赖, 高版本的vesearch对应的glibc也较高,可能需要重新编译新版本的zlib之类的库(如有不会调试的,欢迎骚扰!)
安装完后,将指定安装目录加入系统环境,集群或超算建议使用共享目录,多节点同时运行,方便后面直接运行,安装好后可查看版本:
3、vsearch分析步骤:按顺序
###双端配对,使用参数mergepairs ,与usearch使用方法一致,但注意加参数的时候的格式
vsearch --fastq_mergepairs fastq_1.fq --reverse fastq_2.fq --fastqout merged.16s1.fq --relabel @
# label可以按自己喜好,但要注意与后面的label提取对应,一般不建议修改
merge 结果: 注意merged后面的百分数,正常应该比较高,如果远低于其他文献或者自己其他样品,需要注意 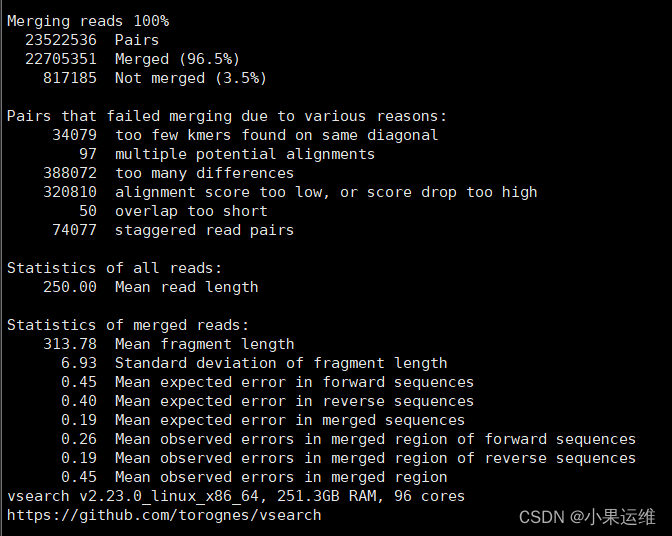
翻转序列,并与原序列合并:
###翻转序列,并将翻转序列与原序列合并到一个文件
vsearch --fastx_revcomp merged.16S1.fq --fastqout merged.16S1_rc.fq
#多个文件可使用for语句#翻转完成后直接合并原序列
cat merged.16S1.fq merged.16S1_rc.fq >mergedFR.16s1.fq
#或
cat merged.16S1{,_rc}.fq>mergedFR.16s1.fq
###合并后查看文件大小是否为原来两倍大小使用python脚本fastq_strip_barcode_relabel2.py提取对应barcode的序列,并重新标记label为16s
python脚本参考,大家可自行到usearch或其他地方下载:扩增子分析中需要使用到的python脚本资源-CSDN文库
###注意python需Python2环境,脚本位置,barcode序列(这里用的是16S其中的典型序列之一,以及样品barcode文件,文件格式间下方:
python /py/fastq_strip_barcode_relabel.py mergedFR.16S1.fq GTGCCAGCMGCCGCGGTAA barcode.txt B16s > barcode.relabel.16S1.fq###barcode.txt 格式
>F_2
AGTTCATACGGC
>F_3
TCGCTTTAACCT
>F_4
基于barcode分离出的样品序列单独再次翻转,并加上label后缀
###
vsearch --fastx_revcomp barcode.relabel.16S1.fq --label_suffix _RC --fastqout barcode.relabeled.16S1_rc.fq再利用反向barcode提取分样:
###这里的反向barcode特征序列和样品barcode按自己实际替换。
python /nfs/sopt/py/fastq_strip_barcode_relabel2.py barcode.relabeled.16S1_rc.fq GGACTACHVGGGTWTCTAAT barcode_16S_r2.txt B16s > mergedFR.relabeled2.16S1.fq将同一批不重复样品的所有正反分样的序列合并到一起进行otu分析和物种分类
###合并所有已标记样品名称的序列
cat mergedFR.relabeled2.16S1.fq mergedFR.relabeled2.16S1.fq {...} > mergedFR.relabel.16s.fq###fastq过滤,去除读长较短的序列
vsearch --fastq_filter mergedFR.relabel.16s.fq --fastq_maxee 0.5 --fastq_minlen 250 --fastq_trunclen 250 --fastq_maxns 1 --fastaout mergedFR.relabel.16S.QC.fa###获取无重复序列unique_seqs
vsearch --derep_fulllength mergedFR.relabel.16S.QC.fa --sizeout --relabel Uniq --output unique_seqs.fa###unique序列排序,加速后续分析
vsearch --sortbysize unique_seqs.fa --output sorted.16s.fa --minsize 2###使用unoise3处理输出otu序列和tab表,新版本特性
###现在版本的vsearch还是alpha版本,所以先用usearch开放版本处理
usearch -unoise3 sorted.16s.fa -zotus zotus.fa -tabbedout uniose3.txt###同样使用usearch开放版本处理uniose3聚类模块,获取otutable
usearch -unoise3 unique_seqs.fa -zotus ref_zotus.fa -minsize 9
usearch -otutab mergedFR.relabel.16S.QC.fa -zotus zotus.fa -otutabout otu_table_16S_unoise3.txt###同样可以使用vsearch的usearch-global模块获取数据otu丰度表
vsearch --usearch_global mergedFR.relabel.16S.QC.fa --db zotus.fa --id 0.99 --otutabout otus_counts.txt###使用rdp数据库的classifier进行物种分类,可按服务器实际资源调整内存
java -Xmx8g -jar /rdp_classifier_2.12/dist/classifier.jar classify -c 0.5 -f filterbyconf -o classification.filterbyconf.16s.txt zotus.fa
以下是私房菜,全vsearch分析流程,可放入脚本直接运行,敬请收藏:
###python脚本环境需要py2,使用前可以先使用conda激活conda环境,或者直接在py2环境下运行
###序列文件,barcode及特征序列请根据自己实际修改;vsearch --version
echo ---------------------------------------------
date
echo Mergepairs and relabel with "@"
vsearch --fastq_mergepairs ./datalink/fastq_1.fq \--reverse ./datalink/fastq_2.fq \--fastqout a.merged.fq \--relabel @
echo Mergepairs over!
echo ---------------------------------------------
date
echo ---------------------------------------------
vsearch --fastx_revcomp a.merged.fq \--label_suffix _RC \--fastqout a.merged_rc.fq
echo ---------------------------------------------
date
echo ---------------------------------------------
cat a.merged.fq a.merged_rc.fq > a.mergedFR.fq
echo --------------------------------------------
python ./testlink/py/fastq_strip_barcode_relabel2.py a.mergedFR.fq \GGACTACHVGGGTWTCTAAT ./datalink/barcode_16S.txt B16S > b.barcode.16S.fq
echo Barcode_16S over!
echo ---------------------------------------------
date
echo ---------------------------------------------
echo Revcomp 16s start
vsearch --fastx_revcomp b.barcode.16S.fq \--fastqout c.barcode.16S_rc.fq
echo Revcomp 16s over!
echo ---------------------------------------------
date
echo ---------------------------------------------
cat b.barcode.16S.fq c.barcode.16S_rc.fq > c.barcode.16S_FR.fqecho Fastq filter start!
vsearch --fastq_filter c.barcode.16S_FR.fq \--fastq_maxee 0.5 \--fastq_minlen 250 \--fastq_trunclen 250 \--fastq_maxns 1 \--fastaout d.barcode.16S_FR.QC.fa
echo Fastq filter over!
echo ---------------------------------------------
date
echo ---------------------------------------------
echo Derep start! Dereplicate across samples and remove singletons.
vsearch --derep_fulllength d.barcode.16S_FR.QC.fa \--output e.dereped.16S.fa \--sizeout
echo Derep over!
echo ---------------------------------------------
date
echo ---------------------------------------------
echo Sortbysize!
vsearch --sortbysize e.dereped.16S.fa \--output f.sorted.16S.fa \--minsize 2
echo ---------------------------------------------
echo Cluster_size start! Precluster at 97% before chimera detection.
vsearch --cluster_size f.sorted.16S.fa \--id 0.97 \--strand plus \--sizein \--sizeout \--relabel OTU_ \--uc g.cluster_size.16S.uc \--centroids g.cluster_size.16S.fa
echo Cluster_size over!
echo ---------------------------------------------
date
echo ---------------------------------------------
echo De novo chimera detection.
vsearch --uchime_denovo g.cluster_size.16S.fa \--sizein \--sizeout \--nonchimeras h.denovo.nonchimeras.16S.fa
echo Obtained unique sequences after de novo chimera detection.
echo ---------------------------------------------
date
echo ---------------------------------------------
echo Usearch_global work start!
vsearch --usearch_global d.barcode.16S_FR.QC.fa \--db h.denovo.nonchimeras.16S.fa \--strand plus \--id 0.97 \--maxaccepts 4 \--maxrejects 128 \--uc i.map_rdp_16s.uc
echo Global over!
date
echo ---------------------------------------------
echo Convert .uc to .txt
python ./testlink/py/uc2otutab.py i.map_rdp_16s.uc > j.OTU_table_16S.txt
echo Convert over!
date
echo ---------------------------------------------
echo Start RDP classify!
java -Xmx200g \-jar /rdp_classifier_2.12/dist/classifier.jar classify \-c 0.5 \-f filterbyconf \-o k.class.filterbyconf.16S.txt h.denovo.nonchimeras.16S.fa
echo RDP Classify work over!
date
echo All 16S sequences processes done!
有不足支出敬请指正!!
这篇关于使用vsearch进行16s扩增子高通量序列分析步骤的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!






