本文主要是介绍孟德尔随机化+WGCNA+预后模型,7+轻松get,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
今天给同学们分享一篇生信文章“Exploring the causality and pathogenesis of systemic lupus erythematosus in breast cancer based on Mendelian randomization and transcriptome data analyses”,这篇文章发表在Front Immunol期刊上,影响因子为7.3。

结果解读:
SNP的选择
总体而言,这项MR研究分析了共计243,218名欧洲血统个体(128,178例患者和115,040例对照组)以及107,936名东亚血统人群(9,774例患者和98,162例对照组)。作者从GWAS中提取了与SLE显著相关的IVs(p < 5 × 10 −8 ),并去除了LD(r 2 <0.001,10,000-kb)。此外,作者分析中的F统计量大于100,表明这些IVs能够强有力地预测SLE的发生率。
遗传易感性与SLE和乳腺癌风险
MR分析显示欧洲队列中SLE与乳腺癌之间不存在因果关联(乳腺癌:OR 0.9985,95%CI 0.9873-1.0099,p=0.79;ER+乳腺癌:OR 0.9974,95%CI 0.9850-1.0101,p=0.69;ER-乳腺癌:OR 1.009,95%CI 0.99-1.02,p=0.22)。没有证据表明其他MR方法基于乳腺癌风险增加。然而,注意到东亚人群中SLE和乳腺癌的遗传易感性的因果推断(IVW:OR:0.95,95%CI:0.92-0.98,p=0.006;加权中位数:OR:0.93,95%CI:0.88-0.97,p=0.002;MR-PRESSO:OR 0.95,95%CI:0.92-0.98,p=0.004)(图2)。多变量MR分析也支持了SLE与东亚人群中乳腺癌显著相关的发现(SNPs:25,OR:0.95,95%CI:0.92-0.98,p=0.0013),在调整混杂因素(吸烟,特征ID:ieu-b-4877)后。
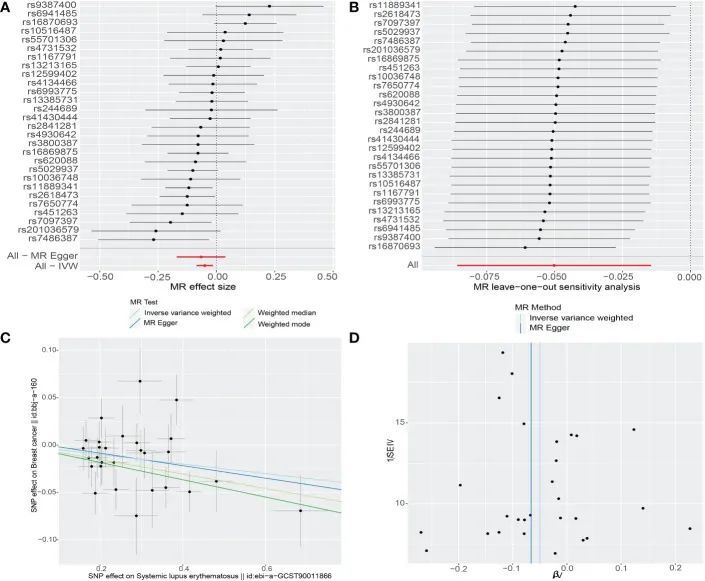
MR估计的敏感性分析
首先,作者进行了MR-Egger回归分析以研究水平多效性,结果表明多效性不太可能对因果关系产生偏倚(所有p值>0.05)。其次,MR-PRESSO测试的结果与无异常值的IVW方法一致,表明原始结果可靠。第三,考虑到东亚队列中SLE和乳腺癌之间的潜在关系,作者进行了逐一排除分析和Cochrane Q检验。逐一排除分析发现没有单个SNP驱动SLE和乳腺癌之间的因果关系(图2)。Cochrane Q检验的p值均大于0.05(IVW测试的Q值为32.93,p=0.2;MR-Egger测试的Q值为32.8,p=0.17),表明SNP之间没有异质性。
识别乳腺癌患者中与SLE相关的差异表达基因
在标准化微阵列结果之后(图3A、B),识别出SLE和乳腺癌相关数据集之间的447个共同差异表达基因(图3C)。通过聚类,WGCNA(软阈值功率=6)进一步去除了灰色模块中的61个明显异常值,并识别出386个感兴趣的关键基因(图3D、E)。

功能特性分析
为了进一步了解乳腺癌中386个SLE-DEGs的潜在功能,作者进行了GO和KEGG富集分析。GO分析显示DEGs富集在细胞周期、细胞增殖和对激素的反应方面(图3F)。KEGG富集分析主要涉及与癌症和细胞周期相关的途径,包括代谢途径、微小RNA、转录调控失调、蛋白聚糖和中心碳代谢(图3G)。
PPI网络和关键基因分析
首先,对386个常见的DEGs构建了PPI网络。其次,使用Cytoscape的Cytuhubba插件计算了前20个关键基因(AURKA,UBE2C,CDC20,PTTG1,CCNB2,MELK,NDC80,CENPF,PRC1,KIF23,TOP2A,RACGAP1,NUSAP1,HMMR,ASPM,KIF15,TTK,DLGAP5,CCNA2和NCAPG)(图4A)。第三,斯皮尔曼相关分析显示这二十个关键基因之间存在显著密切的关联(所有p值<0.0001)(图4B)。
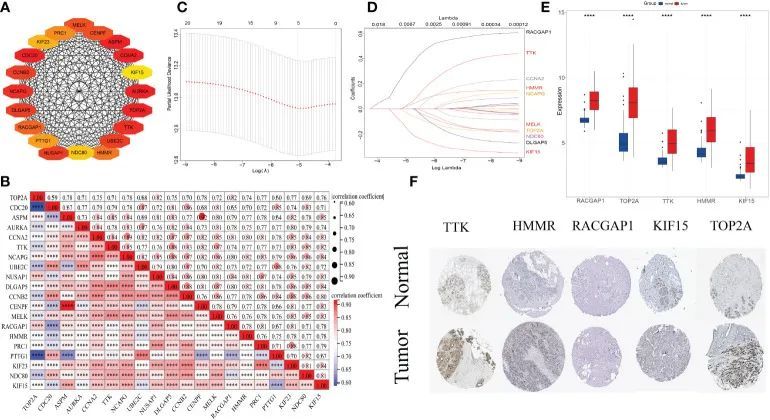
建立和验证预测模型
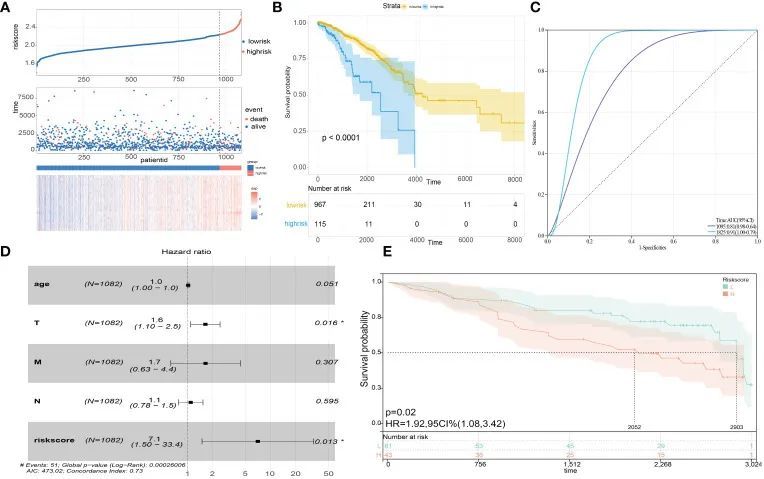
LASSO回归方法被用于优化20个关键基因。最终,选择了最有价值的五个预测基因(RACGAP1、HMMR、TTK、TOP2A和KIF15)来构建SLEscore(图4C、D)。SLEscore的计算方法如下:[-0.036 × TOP2A的表达值] + [0.032 × TTK的表达值] + [0.32 × RACGAP1的表达值] + [0.024 × HMMR的表达值] + [-0.16 × KIF15的表达值]。SLEscore的所有五个关键基因在肿瘤样本中均显著上调(图4E-G)。根据SLEscore,乳腺癌患者被分为两个亚组,其中SLEscore high 与五个预后分子的较高表达水平相关(图5A)。与低SLEscore组相比,高SLEscore组与明显更差的总生存期相关(图5B)。ROC曲线表明,SLEscore可以作为预测乳腺癌患者总生存期的敏感标志物(3年AUC:0.81,5年AUC:0.91)(图5C)。此外,多变量COX回归分析表明,SLEscore是乳腺癌患者的独立风险因素(HR 7.1,95%CI 1.50-33.4,p=0.013)(图5D)。作者建立的SLEscore的C指数为0.73(标准误差:0.043)。在GSE42568数据集中进一步验证了SLEscore,表明利用TCGA数据库构建的SLEscore是乳腺癌患者的独立预后因子(HR 1.92,95%CI 1.08-3.42,p=0.02)(图5E)。随后,作者通过整合SLEscore、年龄和TNM分期为乳腺癌患者建立了一个预测模型,该模型在预测乳腺癌患者1年、3年和5年的生存率方面表现良好(图6A、B)。
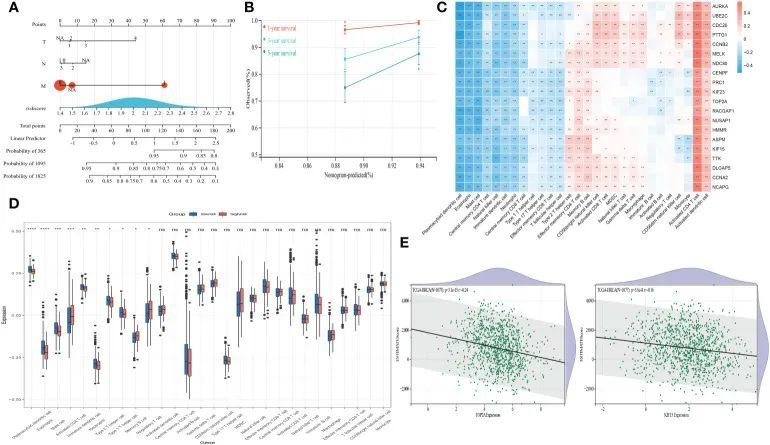
TME细胞的探索,ESTIMATE评分,ICB和PANoptosis
Spearman相关分析揭示了20种分子与TME浸润细胞之间的显著关联(图6C)。值得注意的是,高SLE得分与较低的树突状细胞、嗜酸性粒细胞、肥大细胞、CD4+ T细胞和辅助T细胞表达显著相关(图6D)。ESTIMATE得分与五个关键基因的表达水平呈负相关(所有p值<0.05)(图6E),表明其与疾病结果和肿瘤浸润免疫环境的关系。SLE得分与八种ICB和PANoptosis基因模式显著相关,这些已被证明是乳腺癌患者的预后生物标志物。
总结
作者的MR分析表明,在东亚人群中,SLE患者对乳腺癌症的风险较低。本研究还为癌症和SLE患者的分层提供了路线图,有助于改善个性化随访和个性化决策的策略。
这篇关于孟德尔随机化+WGCNA+预后模型,7+轻松get的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!







