本文主要是介绍你知道红细胞基因对单细胞分析的影响吗,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
大家好,今天周日。最近发现有些单细胞测序数据结果不是很好,或许在作者取样的时候,就注定了后续的生信分析不会太成功~
本次主要发现一个数据集中出现一大群红细胞基因高表达亚群,对后续分析影响还是挺大的。下面先介绍一下为啥单细胞测序前,我们通常要去除红细胞
1 单细胞测序之前的样本处理流程
下图是单细胞测序之前的样本处理流程
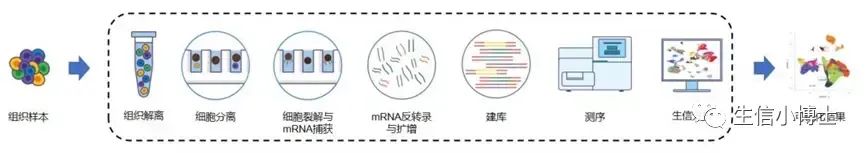
对于组织解离出来的细胞悬液,其质控主要采取对细胞悬液进行台盼蓝染色观察为主。若组织块消化完全,显微镜下观察细胞无成团或聚集现象,细胞悬液即为达标;同时单细胞实验要求细胞悬液符合以下标准:①细胞活性>85%;②细胞总数> 20000;③杂质或红细胞占比小于20%
对于离心重悬后的获取到的细胞悬液,如果红细胞占比大于20%,则需要进行红细胞裂解步骤,裂红后细胞悬液需通过镜检判断红细胞是否裂解彻底,若红细胞数量依然大于20%,则需要二次裂红处理;若红细胞占比小于10%可以直接清洗重悬镜检。
2 我们不禁要问下面两个问题:
-
为什么单细胞测序过程中要去除红细胞?
-
如果红细胞没有去除干净,在后续分析时,出现大量红细胞基因高表达亚群咋办?
为什么单细胞测序过程中要去除红细胞:
-
由于红细胞不包含核糖体,其RNA序列主要由血红蛋白基因(HBB)组成,这些序列对于我们研究其他细胞的基因表达没有太多意义,因此会降低其他细胞的RNA测序效率。
-
测序深度的固定:在单细胞测序中,通常会设定一个固定的总测序深度,即总测序的数据量是一定的。如果样本中存在大量的红细胞,它们的RNA序列会占据较大比例的总测序深度,从而减少其他细胞的测序深度。这就意味着其他细胞的RNA序列被稀释了,其表达水平可能无法准确地检测和分析。
在后续分析时,出现大量红细胞基因高表达亚群咋办:
-
数据过滤和筛选:通过对单细胞测序数据进行筛选和过滤,将红细胞基因高表达的细胞排除在分析之外。
-
数据纠正和规范化:使用专门的数据纠正方法,如Scrublet、SoupX等,对红细胞干扰进行更精确的估计和消除。这些方法可以校正红细胞引起的扭曲,减少其对其他细胞的影响。
-
细胞亚群分析:如果红细胞基因高表达的亚群数量较少,可以将其视为一个独立的细胞亚群进行分析。这样可以避免红细胞的影响对其他细胞群体的解读造成干扰。
我发现使用SoupX这个工具比较方便,就三句代码,大家可以去官网自己看看:
#https://github.com/constantAmateur/SoupX#https://rawcdn.githack.com/constantAmateur/SoupX/204b602418df12e9fdb4b68775a8b486c6504fe4/inst/doc/pbmcTutorial.htmlsc = load10X('path/to/your/cellranger/outs/folder')sc = autoEstCont(sc)out = adjustCounts(sc)
但是,
我觉得最好的办法:单细胞测序前尽可能地去除红细胞,以避免这类问题的出现。
最后,祝各位在分析实战过程中,都能得偿所愿,顺利发表~

![]()
看完记得顺手点个“在看”哦!
这篇关于你知道红细胞基因对单细胞分析的影响吗的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!






