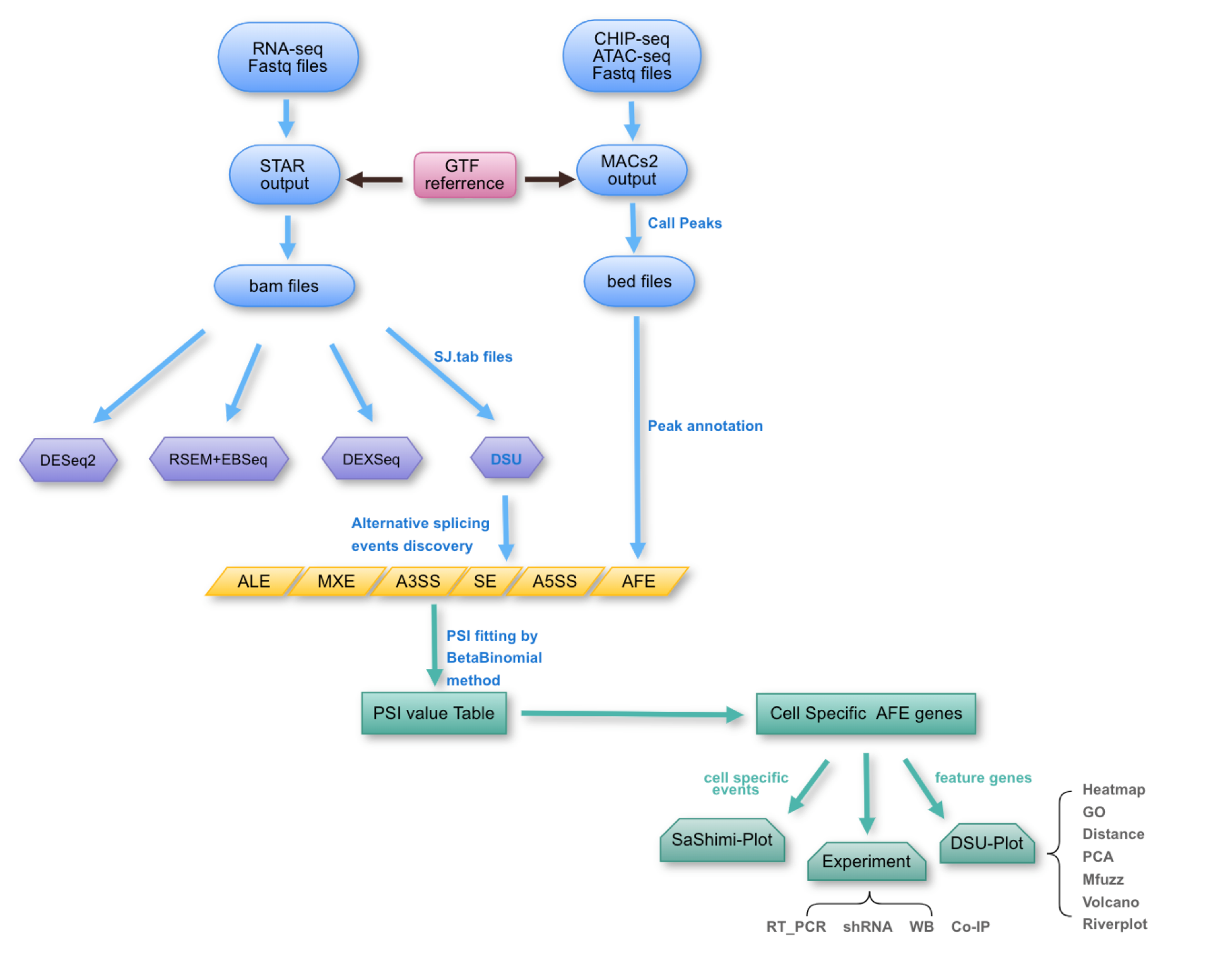
首先拿到二代测序下机数据fastq,STAR软件比对之后我们提取junction counts的文件信息。利用R统计语言中的multiple merge将所有样本的junction counts整合到一个表里面。
下面是自己开发R流程,DSU(目前是bulk数据和singlecell的数据都适用,本流程只用bulk的处理方法。)
得到的table放入R中判断出相同start以及相同end的junction 并将其组合成配对pair的junction counts list,
将不同的剪切事件AFE ALE SE A5SS A3SS MXE 等分辨出来贴上AStype的类型,找出各自的Hostgene(参见上篇GTF的导入以及处理流程)作为相应的注释信息。
本人关注AFE事件,将AFE相关的事件找出来后,利用多种统计学方法寻找其细胞特异性的事件并且指导下游实验,画出sashimiplot、热图、降纬图、火山图、河流图以及clustering图。
其中关键的是有chipseq 以及ATACseq数据的辅助支持,对位点信息的定量统计,以及表观信号的验证。当在rnaseq 数据中有较好的验证(sashimiplot)、chipseq ATACseq较好的验证之后(MACs2 call peak with singals)我们就比较有信心合把握对下游的实验验证进行操作。
shRNA qRTPCR等等。