本文主要是介绍(五)单细胞数据分析——细胞亚群的表型特征刻画(补充工作),希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
由于细胞表型特征这里,我们可以进行一些信息的补充,一个是从全局重新观察每个细胞亚群的基因表达,另一个是细胞亚群特征基因的小提琴图。
第一步:导入R包
library(scibet) library(Seurat) library(scater) library(scran) library(dplyr) library(Matrix) library(cowplot) library(ggplot2) library(harmony)
第二步:全局观察每个细胞亚群的基因表达
导入所有细胞亚群的数据
BRCA_SingleCell.Bcell <- readRDS("BRCA_SingleCell.B_Pcell.RDS") BRCA_SingleCell.Tcell <- readRDS("BRCA_SingleCell.Tcell.RDS") BRCA_SingleCell.Mycell <- readRDS("BRCA_SingleCell.Mycell.RDS") BRCA_SingleCell.Fcell <- readRDS("BRCA_SingleCell.Fcell.RDS")设置颜色种类
cb_palette <- c("antiquewhite", "yellowgreen", "thistle", "violet", "azure3", "azure4", "black","blue", "blue4","blueviolet", #B"brown4","brown1", "burlywood1", "burlywood4", "cadetblue1", "cadetblue4","chartreuse", "chartreuse3", #T"chartreuse4","chocolate1", "chocolate3", "chocolate4", "cyan3", "darkgoldenrod","darkgoldenrod1","darkgoldenrod4", "darkolivegreen", "khaki", "yellow" ,"hotpink" ,"hotpink4" , #My"slateblue1" ,"slateblue4", "yellow3","sienna4", "tomato1", "sienna1", "dodgerblue4", "darkmagenta","cornsilk4") #F绘制每个细胞亚群的UMAP图
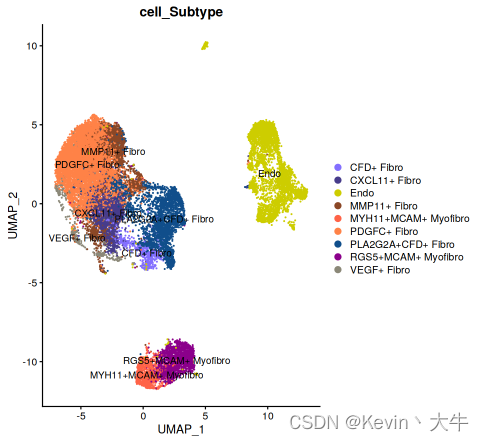
DimPlot(object = BRCA_SingleCell.Bcell,reduction = 'umap',group.by = c('cell_Subtype'),cols = cb_palette[1:10], label = TRUE) DimPlot(object = BRCA_SingleCell.Tcell,reduction = 'umap',group.by = c('cell_Subtype'),cols = cb_palette[11:18], label = TRUE) DimPlot(object = BRCA_SingleCell.Mycell,reduction = 'umap',group.by = c('cell_Subtype'),cols = cb_palette[19:31], label = TRUE) DimPlot(object = BRCA_SingleCell.Fcell,reduction = 'umap',group.by = c('cell_Subtype'),cols = cb_palette[32:40], label = TRUE)其中一个结果如下:
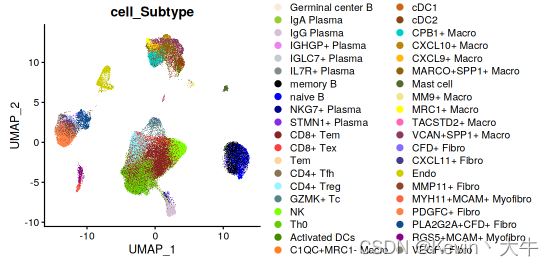
合并所有数据绘制总的UMAP图
BRCA_SingleCell <- merge(BRCA_SingleCell.Fcell,BRCA_SingleCell.Tcell) BRCA_SingleCell <- merge(BRCA_SingleCell,BRCA_SingleCell.Bcell) BRCA_SingleCell <- merge(BRCA_SingleCell,BRCA_SingleCell.Mycell) DimPlot(object = BRCA_SingleCell,reduction = 'umap',group.by = c('cell_Subtype'), cols = cb_palette[1:40], label = FALSE)结果如下:
分析不同细胞亚群之间的差异基因,并绘制热图
cellType_markerGene <- findMarker(BRCA_SingleCell) top <- cellType_markerGene %>% group_by(cluster) %>% top_n(n =10, wt = myAUC) top$cluster %>% levels(.) clusters <- unique(BRCA_SingleCell$cell_Subtype) genes <- c() for(i in clusters){gene <- as.vector(as.matrix(subset(top, cluster == i, gene)))gene <- gene[1:(ifelse(length(gene)>5,5,length(gene)))]names(gene) <- rep(i, times = length(gene))genes <- c(genes, gene) } table(names(genes)) table(unique(names(genes))) B_P_gene <- c(genes[which(names(genes) == "naive B")],genes[which(names(genes) == "Memory B")],genes[which(names(genes) == "Germinal center B cell")],genes[which(names(genes) == "IgA Plasma")],genes[which(names(genes) == "IGHGP+ Plasma")],genes[which(names(genes) == "IGLC7+ Plasma")],genes[which(names(genes) == "IL7R+ Plasma")],genes[which(names(genes) == "NKG7+ Plasma")],genes[which(names(genes) == "IgG Plasma")]) T_gene <- c(genes[which(names(genes) == "CD8+ Tem")],genes[which(names(genes) == "CD4+ Tfh")],genes[which(names(genes) == "NK")],genes[which(names(genes) == "Th0")],genes[which(names(genes) == "GZMK+ Tc")],genes[which(names(genes) == "CD8+ Tex")],genes[which(names(genes) == "CD4+ Treg")],genes[which(names(genes) == "Tem")]) Stroma_gene <- c(genes[which(names(genes) == "MYH11+MCAM+ Myofibro")],genes[which(names(genes) == "RGS5+MCAM+ Myofibro")],genes[which(names(genes) == "VEGF+ Fibro")],genes[which(names(genes) == "CFD+ Fibro")],genes[which(names(genes) == "PLA2G2A+CFD+ Fibro")],genes[which(names(genes) == "PDGFC+ Fibro")],genes[which(names(genes) == "CXCL11+ Fibro")],genes[which(names(genes) == "MMP11+ Fibro")],genes[which(names(genes) == "MMP1+ Fibro")],genes[which(names(genes) == "MMP11+ Fibro")]) Myeloid_gene <- c(genes[which(names(genes) == "MM9+ Macro")],genes[which(names(genes) == "C1QC+MRC1- Macro")],genes[which(names(genes) == "MRC1+ Macro")],genes[which(names(genes) == "cDC2")],genes[which(names(genes) == "VCAN+SPP1+ Macro")],genes[which(names(genes) == "MARCO+SPP1+ Macro")],genes[which(names(genes) == "cDC1")],genes[which(names(genes) == "Activated DCs")],genes[which(names(genes) == "Mast cell")],genes[which(names(genes) == "TACSTD2+ Macro")],genes[which(names(genes) == "CXCL10+ Macro")]) cellMarker <- c(B_P_gene,T_gene,Myeloid_gene,Stroma_gene) length(cellMarker) length(unique(cellMarker)) sum(table(cellMarker) == 1) cellMarker.unique <- unique(cellMarker) options(repr.plot.height = 14, repr.plot.width = 18) test <- DotPlot(object = BRCA_SingleCell,features = cellMarker.unique,cols = c('blue','red'),group.by = 'cell_Subtype')+ theme_bw()+ theme(axis.text.x = element_text(angle = -90,hjust = 0,vjust = 0)) + coord_flip() test test$data$id %>% unique(.) %>% setdiff(.,c('naive B','IL7R+ Plasma','memory B','Germinal center B','IGHGP+ Plasma','IGLC7+ Plasma','NKG7+ Plasma','STMN1+ Plasma','IgA Plasma','IgG Plasma','Th0','CD4+ Tfh','CD8+ Tem','CD4+ Treg','GZMK+ Tc','CD8+ Tex','NK','Tem','TACSTD2+ Macro','MARCO+SPP1+ Macro','VCAN+SPP1+ Macro','CXCL10+ Macro','cDC2','CXCL9+ Macro','MRC1+ Macro','cDC1','Activated DCs','C1QC+MRC1- Macro','MM9+ Macro','CPB1+ Macro','Mast cell','CXCL11+ Fibro','CFD+ Fibro','RGS5+MCAM+ Myofibro','MMP11+ Fibro','VEGF+ Fibro','PLA2G2A+CFD+ Fibro','MYH11+MCAM+ Myofibro','PDGFC+ CAFs','Endo')) Mmarker_data <- test$data Mmarker_data$Zscore <- Mmarker_data$avg.exp.scaled Mmarker_data$cluster <- factor(x = Mmarker_data$id,levels = c('naive B','IL7R+ Plasma','memory B','Germinal center B','IGHGP+ Plasma','IGLC7+ Plasma','NKG7+ Plasma','STMN1+ Plasma','IgA Plasma','IgG Plasma','Th0','CD4+ Tfh','CD8+ Tem','CD4+ Treg','GZMK+ Tc','CD8+ Tex','NK','Tem','TACSTD2+ Macro','MARCO+SPP1+ Macro','VCAN+SPP1+ Macro','CXCL10+ Macro','cDC2','CXCL9+ Macro','MRC1+ Macro','cDC1','Activated DCs','C1QC+MRC1- Macro','MM9+ Macro','CPB1+ Macro','Mast cell','CXCL11+ Fibro','CFD+ Fibroblast','RGS5+MCAM+ Myofibro','MMP11+ Fibro','VEGF+ Fibro','PLA2G2A+CFD+ Fibro','MYH11+MCAM+ Myofibro','PDGFC+ Fibro','Endo'),labels = c('naive B','IL7R+ Plasma','memory B','Germinal center B','IGHGP+ Plasma','IGLC7+ Plasma','NKG7+ Plasma','STMN1+ Plasma','IgA Plasma','IgG Plasma','Th0','CD4+ Tfh','CD8+ Tem','CD4+ Treg','GZMK+ Tc','CD8+ Tex','NK','Tem','TACSTD2+ Macro','MARCO+SPP1+ Macro','VCAN+SPP1+ Macro','CXCL10+ Macro','cDC2','CXCL9+ Macro','MRC1+ Macro','cDC1','Activated DCs','C1QC+MRC1- Macro','MM9+ Macro','CPB1+ Macro','Mast cell','CXCL11+ Fibro','CFD+ Fibro','RGS5+MCAM+ Myofibro','MMP11+ Fibro','VEGF+ Fibro','PLA2G2A+CFD+ Fibro','MYH11+MCAM+ Myofibro','PDGFC+ Fibro','Endo')) unique(Mmarker_data$id) Mmarker_data$geneClass <- NA Mmarker_data$geneClass <- ifelse(Mmarker_data$features.plot %in% B_P_gene,'B',Mmarker_data$geneClass) Mmarker_data$geneClass <- ifelse(Mmarker_data$features.plot %in% T_gene,'T',Mmarker_data$geneClass) Mmarker_data$geneClass <- ifelse(Mmarker_data$features.plot %in% Myeloid_gene,'M',Mmarker_data$geneClass) Mmarker_data$geneClass <- ifelse(Mmarker_data$features.plot %in% Stroma_gene,'S',Mmarker_data$geneClass) library(aplot) options(repr.plot.height = 14, repr.plot.width = 13) group <- factor(c('naive B','IL7R+ Plasma','memory B','Germinal center B','IGHGP+ Plasma','IGLC7+ Plasma','NKG7+ Plasma','STMN1+ Plasma','IgA Plasma','IgG Plasma','Th0','CD4+ Tfh','CD8+ Tem','CD4+ Treg','GZMK+ Tc','CD8+ Tex','NK','Tem','TACSTD2+ Macro','MARCO+SPP1+ Macro','VCAN+SPP1+ Macro','CXCL10+ Macro','cDC2','CXCL9+ Macro','MRC1+ Macro','cDC1','Activated DCs','C1QC+MRC1- Macro','MM9+ Macro','CPB1+ Macro','Mast cell','CXCL11+ Fibro','CFD+ Fibro','RGS5+MCAM+ Myofibro','MMP11+ Fibro','VEGF+ Fibro','PLA2G2A+CFD+ Fibro','MYH11+MCAM+ Myofibro','PDGFC+ Fibro','Endo'),levels = c('naive B','IL7R+ Plasma','memory B','Germinal center B','IGHGP+ Plasma','IGLC7+ Plasma','NKG7+ Plasma','STMN1+ Plasma','IgA Plasma','IgG Plasma','Th0','CD4+ Tfh','CD8+ Tem','CD4+ Treg','GZMK+ Tc','CD8+ Tex','NK','Tem','TACSTD2+ Macro','MARCO+SPP1+ Macro','VCAN+SPP1+ Macro','CXCL10+ Macro','cDC2','CXCL9+ Macro','MRC1+ Macro','cDC1','Activated DCs','C1QC+MRC1- Macro','MM9+ Macro','CPB1+ Macro','Mast cell','CXCL11+ Fibro','CFD+ Fibro','RGS5+MCAM+ Myofibro','MMP11+ Fibro','VEGF+ Fibro','PLA2G2A+CFD+ Fibro','MYH11+MCAM+ Myofibro','PDGFC+ Fibro','Endo')) %>% as.data.frame() %>% mutate(group=c(rep("B cell",10),rep("T cell",8),rep("Myeloid",13),rep("Strom cell",9))) %>%mutate(p="group") %>%ggplot(aes(.,y=p,fill=group))+geom_tile() + scale_y_discrete(position="right") +theme_minimal()+xlab(NULL) + ylab(NULL) +theme(axis.text.x = element_blank())+labs(fill = "Group") heatmap <- ggplot(Mmarker_data, aes(y=features.plot, x=cluster, fill=Zscore))+ geom_raster()+scale_fill_gradient2(low="#003366", high="#990033", mid="white",midpoint = 0)+ theme(axis.text.x = element_text(angle = -90,hjust = 0,vjust = 0.5, size = 15),axis.ticks.y = element_blank())+ xlab(NULL) + ylab(NULL)+theme(panel.border = element_rect(fill=NA,color="black", size=1, linetype="solid"))+geom_vline(xintercept=c(-0.5,10.5,18.5,31.5),size=.8)+geom_hline(yintercept=c(-0.5,18.5,48.5,76.5), size=.8) heatmap <- heatmap %>% insert_top(group ,height = 0.05) heatmap结果如下:


第三步:绘制特征基因的小提琴图
构造绘制小提琴图的函数与特征基因筛选函数
modify_vlnplot<- function(obj, feature, pt.size = 0, plot.margin = unit(c(-0.75, 0, -0.75, 0), "cm"),...) {p<- VlnPlot(obj, features = feature, pt.size = pt.size,group.by = 'cell_Subtype', ... ) + xlab("") + ylab(feature) + ggtitle("") + theme(legend.position = "none", axis.text.x = element_blank(), axis.ticks.x = element_blank(), axis.title.y = element_text(size = rel(1), angle = 0), axis.text.y = element_text(size = rel(1)), plot.margin = plot.margin ) #+ rotate_x_text() return(p) } extract_max<- function(p){ymax<- max(ggplot_build(p)$layout$panel_scales_y[[1]]$range$range)return(ceiling(ymax)) }## main function StackedVlnPlot<- function(obj, features,pt.size = 0, plot.margin = unit(c(-0.75, 0, -0.75, 0), "cm"),...) {plot_list<- purrr::map(features, function(x) modify_vlnplot(obj = obj,feature = x, ...))# Add back x-axis title to bottom plot. patchwork is going to support this?# rotate_x_text() 转置横坐标的字体,ggpubr 包,在这里添加才会只出现一次横坐标,不会出现多个横坐标plot_list[[length(plot_list)]]<- plot_list[[length(plot_list)]] +theme(axis.text.x=element_text(), axis.ticks.x = element_line()) + rotate_x_text(angle = 90)# change the y-axis tick to only max value ymaxs<- purrr::map_dbl(plot_list, extract_max)plot_list<- purrr::map2(plot_list, ymaxs, function(x,y) x + scale_y_continuous(breaks = c(y)) + expand_limits(y = y))p<- patchwork::wrap_plots(plotlist = plot_list, ncol = 1)return(p) } findMarker <- function(x){Idents(x) <- 'cell_Subtype'cellType_markerGene <- FindAllMarkers(x,logfc.threshold = 0.5,only.pos = T,test.use = 'roc',min.pct = 0.2)cellType_markerGene <- subset(cellType_markerGene,!grepl(pattern = 'RP[LS]',gene))cellType_markerGene <- subset(cellType_markerGene,!grepl(pattern = 'MT-',gene))top5 <- cellType_markerGene %>% group_by(cluster) %>% top_n(n = 10, wt = myAUC)print(top5)return(cellType_markerGene) }注:查找差异基因是因为,如果没有去了解特征基因,可以通过差异基因进行一个“预见”
绘制小提琴图(以髓系细胞为例)
cellMarker.Mycell <- findMarker(BRCA_SingleCell.Mycell) features<- c("HLA-DOB","HLA-DQA2","HLA-DPB1","LGALS2","CLEC9A","WDFY4","IDO1","CCND1","IRF8","RGCC","SHTN1","BATF3","DNASE1L3","CD1C","XCR1","CADM1","LIMD2","CPNE3") vlnplt <- StackedVlnPlot(obj = BRCA_SingleCell.Mycell, features = features) vlnplt结果如下:
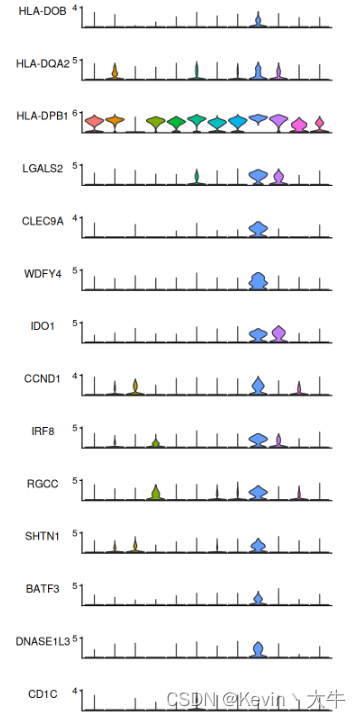
综上所述,补充的工作就完成了,主要是对描述进行一个更细致的刻画,通过热图和总UMAP全局观察细胞亚群,通过小提琴图刻画特征基因在细胞亚群中的表达!
这篇关于(五)单细胞数据分析——细胞亚群的表型特征刻画(补充工作)的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!





