本文主要是介绍4+内质网应激+预后模型教你如何应用到自己的生信分析研究中。,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
今天给同学们分享一篇内质网应激+预后模型的生信文章“Characteristics of endoplasmic reticulum stress in colorectal cancer for predicting prognosis and developing treatment options”,这篇文章于2023年3月31日发表在Cancer Med期刊上,影响因子为4。
越来越多的证据支持内质网应激(ERS)在结直肠癌(CRC)中起着重要作用。在本研究中,作者开发了一个与ERS相关的基因(ERSRGs)模型,以帮助评估和治疗CRC患者的预后。
1. ERSRGs的富集分析
首先,作者在GeneCard数据库中搜索关键词“内质网应激”,以识别一组ERSRGs。使用|log2FC| = 0.585和FDR <0.05作为阈值进行基因表达水平比较,共检测到220个DEGs。然后对DEGs的富集进行分析,GO和KEGG结果显示主要与内质网、内质网腔、ERS、内质网蛋白质加工、免疫相关信号通路和CRC相关。这些结果表明ERS在CRC的发展中起着关键作用。
2. 筛选EERSRGs并构建风险评分模型
对220个差异表达基因进行了单变量Cox回归分析,发现了与预后相关的26个基因(p < 0.05)(图1A)。首先,总结了样本中ERSRGs的体细胞突变情况。在541个带有突变信息的CRC样本中,共有115个ERSRGs发生了突变,突变频率为21.26%(图1B)。ATP2A1、PARGC1A和CNGA3的突变率≥4%,其中ATP2A1的突变率最高,达到了5%。然而,LEP、CXCL1、HAMP、C3orf70、SNCG、UTS2和NOL3在CRC样本中的突变率没有显著变化。重要的是,在与预后相关的26个ERSRGs中存在显著的共同突变特征(图1C)。此外,作者分析了这些ERSRGs的拷贝数变化,并观察到这些基因的拷贝数变化频率存在显著差异,FABP4、TERT、C3orf70和ADIPOQ的拷贝数增加,而NGF、UTS2、GRP和SNCG的拷贝数减少,共同表明ERSRGs的异常表达(图1D、E)。鉴于ERS在CRC中的重要作用,作者试图开发一个ERS预后风险评分模型,以更准确地评估CRC患者的状况。构建风险评分模型所涉及的CRC样本的临床信息如表1所示。然后通过LASSO Cox回归分析筛选出16个基因(DDIT3、TERT、PPARGC1A、ATP2A1、GRP、TRAP1、CD36、CXCL1、TRPV3、UTS2、OGT、NOL3、STC2、BDNF、TIMP1和C3orf70)来构建模型(图1F,G)。根据风险评分的中位数将训练集中的高风险组(n = 270)和低风险组(n = 270)进行划分。验证集以相同的方式划分,主成分分析(PCA)显示该模型能够有效地将CRC样本分为高风险和低风险组(图1H,I)。
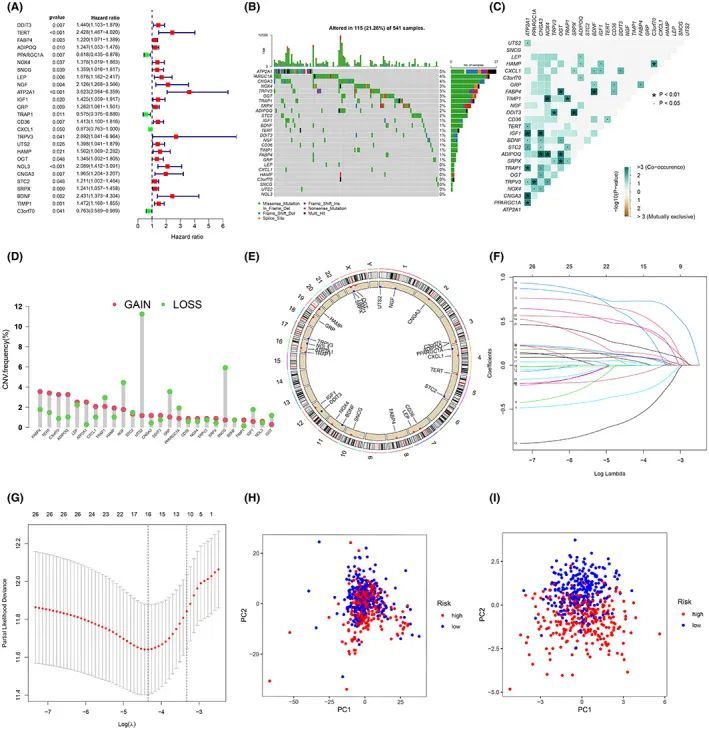
图1 建立ERS风险评分模型
3. 训练集和验证集样本的风险预测和生存状态
从数据集中生成了风险曲线,其中风险评分可视化了两组患者的风险评分(图2A、B)。分析发现高风险组患者的预后较差,随着风险评分的增加,死亡率也增加(图2C、D)。接下来,风险直方图在风险评分模型中直观评估了生存状态。低风险评分的患者生存率显著较高,进一步证明该模型能够准确地分层患者。此外,作者使用热成像技术比较了影响患者预后的ERSRGs在两个风险组之间的表达水平。BDNF、GRP、CD36、TIMP1、DDIT3、OGT、STC2、ATP2A1、NOL3、TERT、TPRV3和UTS2在高风险组中表达水平较高,而PPARGC1A、C3orf70、CXCL1和TARP1的表达水平较低(图2E、F)。作者还发现这些基因之间存在显著关联。
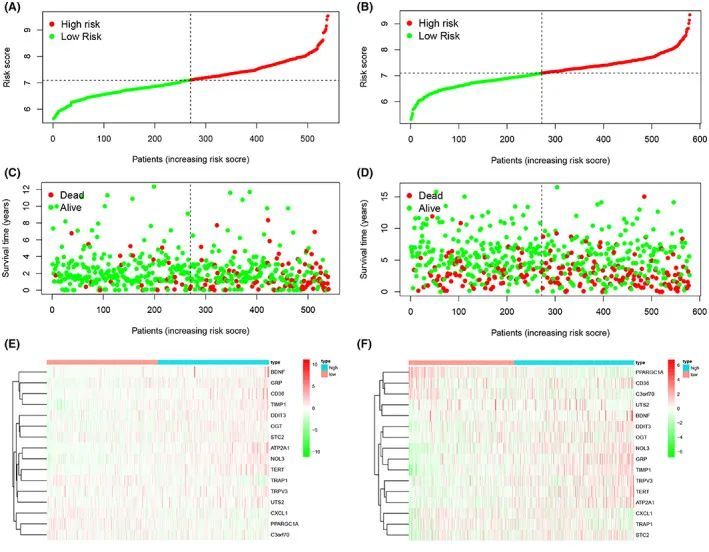
图2 ERS模型中患者的风险预测以及模型中包含的基因表达水平
4. 模型的预测能力
在训练集中,高风险组的OS低于低风险组(p < 0.001)(图3A)。使用ROC曲线验证模型对患者OS的预测效果,1年、3年和5年的AUC结果分别为0.72、0.76和0.77(图3B)。为了检查模型的可靠性,作者在验证集(GSE40967)上进行了测试。高风险组的患者OS较低于低风险组(p < 0.001)(图3C)。验证集的AUC值如图3D所示。为了进一步验证预后模型的准确性,作者在GSE17538数据集中再次验证(p < 0.005)。结果显示高风险组患者的OS较低,并且ROC的AUC值表现良好(图3E,F)。通过计算,作者的模型的R平方约为0.70,这表明作者的预后风险评分模型的预测能力是可接受的。
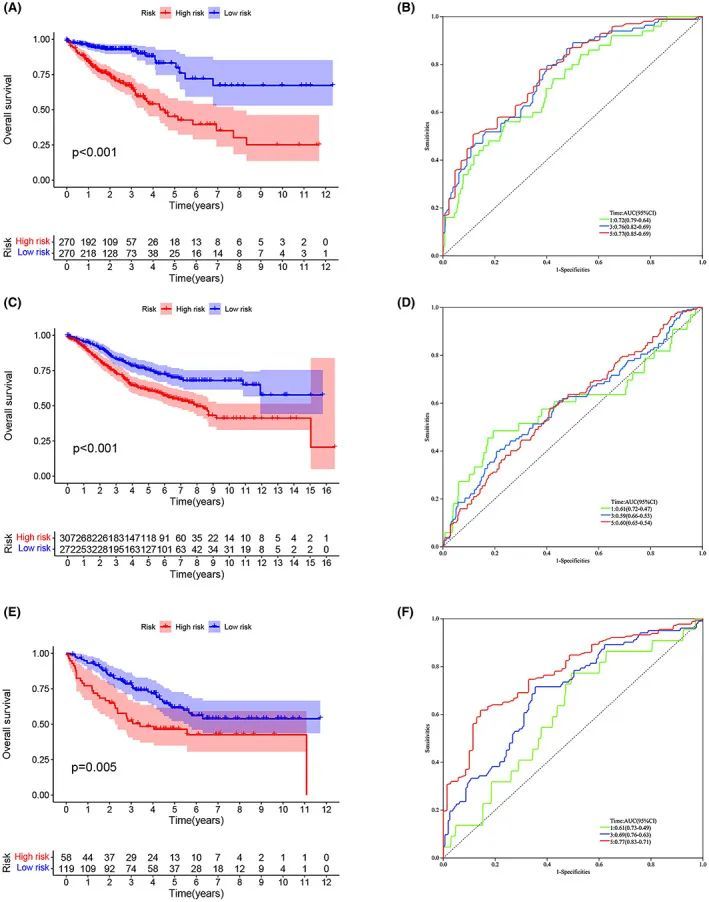
图3 ERS预后风险模型与临床病理特征之间的关系及其对生存的预测价值
5. 预测模型的预测性能比较
为了验证ER应激预后风险评分模型的优越性,通过ROC将之前结直肠癌研究中的模型与作者的模型进行比较。第一个是研究脂质代谢的模型,根据研究者的名字称之为杨氏标志。第二个是研究甲基化的模型,称之为谭氏标志。第三个是研究免疫相关基因的模型,称之为温氏标志。第四个是研究脂肪酸代谢的模型,称之为丁氏标志。根据模型中的基因,作者使用R软件包进行ROC分析。结果显示,作者构建的ERS预后风险评分模型在1年、3年和5年的AUC值均高于其他四个模型(图4A-D)。这意味着作者的风险评分模型具有更好的预测性能。
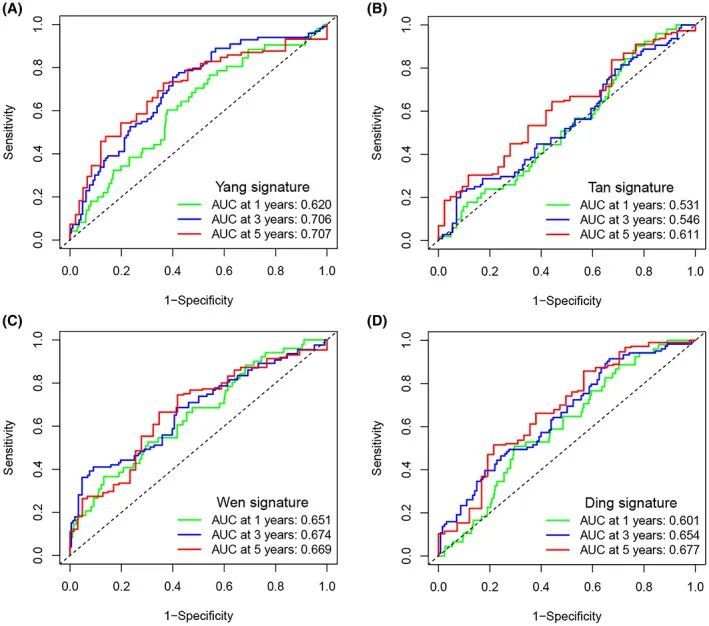
图4 不同预后模型的预测性能
6. 临床病理特征与风险评分的关联
由于不同的临床病理特征对疾病预后有不同的影响,因此还探讨了风险评分模型中临床病理特征的分布情况。结果表明,风险评分与肿瘤进展最为密切相关。尽管风险评分在年龄或性别上没有显著差异(图5A、B),但随着TNM分期和病理分期的升高,风险评分也随之增加(图5C-F)。接下来,比较了风险评分、年龄、性别和病理分期对预后的预测能力,结果显示风险评分的AUC值最高(0.786),表明风险评分具有最佳的预测能力(图5G)。C-指数显示风险评分的表现优于其他临床特征,预测结果的准确性更高(图5H)。图5I、J显示年龄和风险评分是OS的独立预测因子(p < 0.001)。
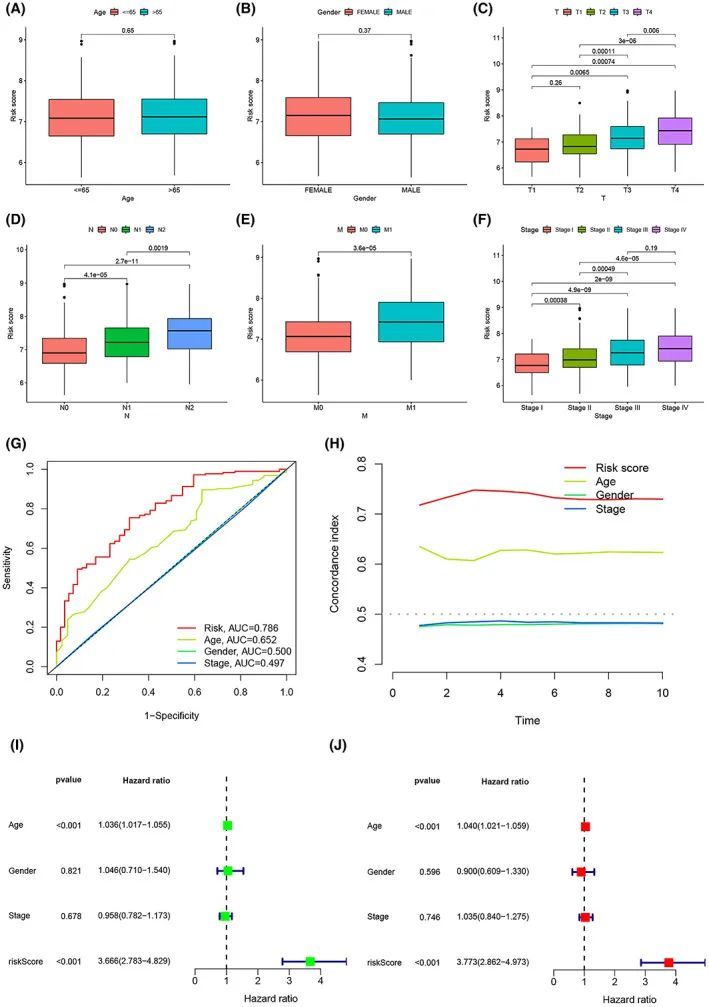
图5 风险评分模型与临床特征之间的关系
7. 构建预测生存率的诺莫图
通过将性别、风险评分、病理分期和年龄从训练集中结合起来,构建了一个预测结直肠癌(OS)的图表模型(图6A)。1年、3年和5年的校准曲线展示了该图表模型的准确性(图6B)。Cox回归分析最终揭示了只有图表模型是独立的预测因子(p < 0.05)(图6C、D)。图6E-G展示了风险评分、图表模型、年龄、性别和病理分期在1年、3年和5年的AUC,其中图表模型的AUC分别为0.780、0.812和0.823,最高(图6H-J)。DCA分析包含了1年、3年和5年的各种临床病理特征。树状图直观地显示了预后风险评分模型的图表模型在1年、3年和5年的DCA曲线中表现最佳。

图6 通过整合训练集中的ERS风险评分和临床病理特征,创建了一个用于预测CRC预后的图表
8. 化疗的反应和相关基因表达的变化
由于结直肠癌(CRC)患者常规在手术后接受化疗,因此探索了模型中患者对化疗药物的反应。使用R软件中的“pRRophetic”分析了风险评分与化疗治疗效果之间的关联。低风险组样本对5-FU的治疗效果较好,而随着风险评分的增加,IC50也增加(图7A、B)。进展无病生存期(PFS)在训练集的亚组之间也显示出显著差异,表明风险评分可以对化疗耐药患者进行分层(p < 0.001)(图7C)。突变型BRAF和TP53以及野生型APC的风险评分较高(图7D-G)。此外,与m6A相关的大多数基因在ERS预后风险评分模型中差异显著,如YTHDF2、YTHDF3、FTO、METTL4、RTCB、GPM6A、SRSF3和CAPRIN1,这些基因可能有助于寻找化疗耐药的治疗靶点(图7H)。
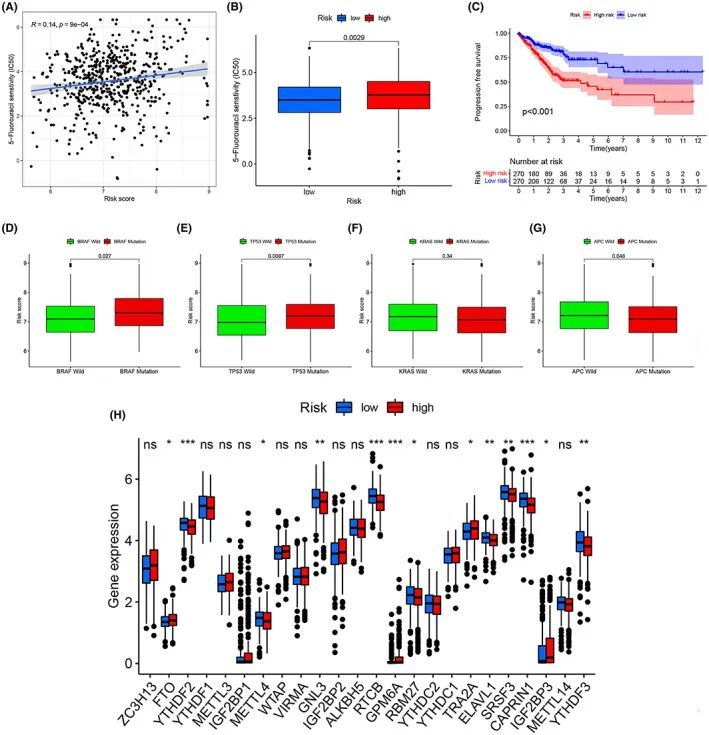
图7 ERS预后风险评分模型在化疗中的作用
9. ERS预测模型中的免疫相关特征
ESTIMATE评分(p < 0.001),免疫评分(p < 0.05)和基质评分(p < 0.001)在两组之间存在显著差异,表明ERS深刻影响免疫和基质细胞浸润(图8A)。图8B、C显示了免疫评分和基质评分与风险评分的相关性。接下来,使用不同软件研究了侵袭性免疫细胞的风险评分。此外,作者发现高风险组中免疫抑制细胞浸润更为丰富,调节性T细胞(Tregs)水平较高,这与高风险组的生存劣势一致,而低水平浸润的T细胞CD4记忆静止(p < 0.001),静止树突状细胞(p < 0.01),活化树突状细胞(p < 0.001)和浆细胞(p < 0.001)(图8D)。在先前的报告中,I型干扰素(IFN-I)信号的激活可以使肿瘤患者从免疫疗法中受益。在本研究中,高风险组中IFN-I反应得到增强,这意味着免疫抑制患者可以通过免疫疗法改善预后(图8E)。为了预测结直肠癌患者对免疫治疗的反应,进一步研究了免疫检查点与内质网应激反应(ERS)之间的相关性(图8F)。作者的研究结果显示,ERS风险评分与大多数免疫检查点呈正相关,如PDCD1(PD-1)、CD274(PD-L1)、CTLA-4、LAG3、TIGIT和HAVCR2(TIM-3)。此外,作者发现高风险组中PD-1、TIGIT、TIM-3和CTLA-4的表达较高。通过免疫治疗队列,探索了ERS风险模型预测结直肠癌预后的能力。结果显示,在没有任何免疫治疗的情况下,高风险组患者对免疫治疗的反应明显较差,并且当仅使用针对CTLA4的免疫治疗时,这一结果仍然存在(图8G,H)。此外,作者获得了训练集的TIDE预测分数,并在ERS风险组之间进行了分析。随着TIDE预测分数的增加,预后结果下降,再次表明低风险组具有更好的预后。接下来,对模型的不同亚组进行免疫抑制基因表达的分析显示,高风险组中大多数免疫抑制基因的表达水平增加,这可能解释了该组患者免疫治疗效果不佳。

图8 ERS预后风险评分模型中的免疫相关特征
10. 模型中差异表达基因的功能富集分析和蛋白质相互作用网络
为了更好地理解基因的作用,对两个风险组的差异表达基因进行了功能富集分析。GO结果显示这些基因参与了包括细胞外区域、细胞外区域分数、结构分子活性、分子功能调节因子、内质网和细胞外结构组织等通路(图9A)。KEGG分析显示了人乳头瘤病毒感染、ECM-受体相互作用、吞噬体、PI3K-Akt信号通路、WNT信号通路、PPAR信号通路和雌激素信号通路等术语的显著富集(图9B)。在线网站STRING用于研究风险组中差异表达基因的蛋白质相互作用,并生成了PPI网络(图S4A)。使用Cytoscape软件对PPI数据进行可视化处理,上调和下调基因的表达分别以红色和绿色标记(图9C)。然后使用Cytoscape的插件cytoHubba对差异表达基因中的中心基因进行筛选,排名前10位如图9D所示。随后比较了正常组织和肿瘤组织中中心基因的差异。结果显示,SPP1、COMP、THBS2、SERPINE1和COL11A1在肿瘤组织中高表达,而MYH11、TAGLN和CNN1低表达。预后价值分析显示,在上述八个具有统计学差异的中心基因中,只有SPP1、TAGLN、COMP、SERPINE1、COL11A1和CNN1的mRNA表达会影响预后(图9E-H),其中SPP1、COMP、SERPINE1和COL11A1的水平与不良预后具体相关。为了验证SPP1、COMP、SERPINE1和COL11A1在正常和CRC组织之间的差异表达,对11名CRC患者的正常和肿瘤活检标本进行了q-PCR检测(图9I-L)。
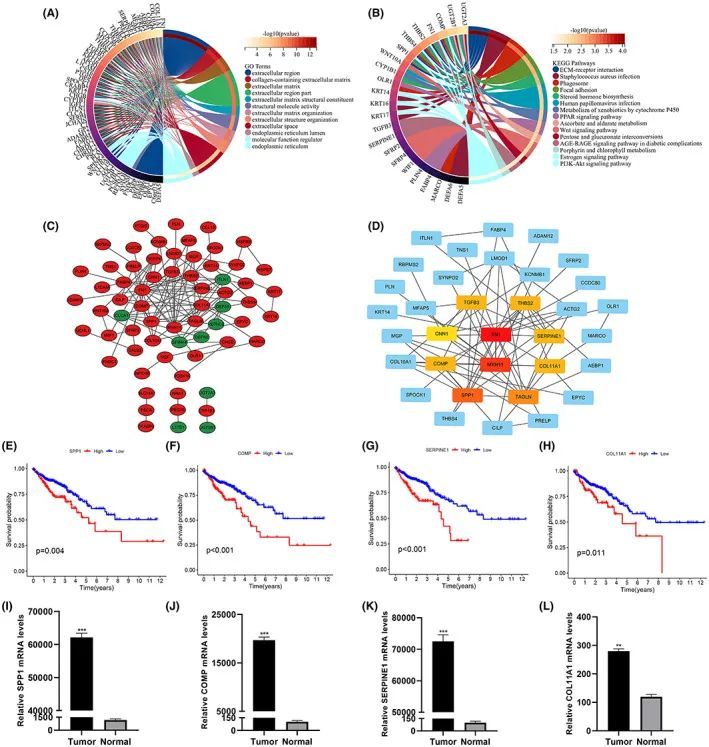
图9 PPI网络的可视化
总结
本研究提供了一种新的、准确的预测CRC患者预后的方案。鉴于作者模型的显著预后能力和准确的治疗结果预测,作者的风险评分模型可以作为患者选择治疗的新工具。考虑到ERS在CRC中的重要性,有必要对其具体作用机制进行更深入的研究。
这篇关于4+内质网应激+预后模型教你如何应用到自己的生信分析研究中。的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!




