genes专题
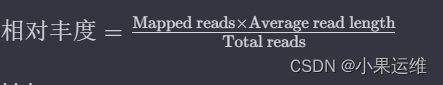
基于BWA,Bowtie2,samtools、checkm等工具计算宏基因组学序列分析中Contigs与Genes在样品中的丰度,多种计算方式和脚本对比
计算contigs和genes相对丰度可以提供有关微生物群落结构和功能的信息。以下是计算这两个指标的意义: 1. Contigs的相对丰度:contigs是利用基因组测序技术获得的碎片序列,通过计算contigs的相对丰度可以了解微生物群落中不同菌种的相对丰度。这可以帮助研究者理解微生物群落的物种组成和群落结构。 2. Genes的相对丰度:基因是生物体内功能的基本单位,通过计算基因的相对丰度

使用机器学习来测量基因间的相关性:一个多特征模型(Using Machine Learning to Measure Relatedness Between Genes)
1. 摘要 测量一对基因间的条件亲缘关系是计算生物学的一项基本技术,也是一个重大的挑战。论文提出了一个新的机器学习模型—多特征相关性(MFR),通过将表达相似度和基于先验知识的相似度纳入评估标准,来准确地测量一对基因之间的条件相关性。 2. 介绍 基因之间的相互作用通常被建模为一对基因之间0/1(非相互作用/相互作用)的二元关系,而亲缘性则意味着一对基因之间的某种程度的关系。

TCGA相关分析之数据筛选 | python从TCGA-GBM的RNA-seq表达数据count中筛选出各genes对应的案例cases的表达量count矩阵
接上一篇文章,现在开始筛选数据组成count矩阵。 上一篇:TCGA下载GBM患者的RNA-seq数据 上一篇结束,下载到初始数据(图一图二是下载之后的文件夹以及每一个文件夹中的count数据文件) 需要从每一个count数据文件中筛选出gene_name、gene_type为lncRNA、FPKM表达量,效果图如下: 由于不会R语言,就用python来实现 步骤: 从每一