atoms专题
MAX/MSP SDK学习03:Atoms and Messages的使用
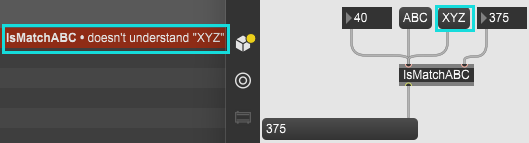
今天终于把Message消息选择器看得有点头绪了,主要是这个官方英文文档理解起来有点抽象。 编写IsMatchABC自定义Object,要求: ①若左入口(入口0)收到 "int" 型消息,则从出口发送数值 "888"; ②若左入口(入口0)收到 "ABC" 消息,则从出口发送 "Message match!!!(Send)"; ③若右入口(入口1)收到 "int" 型消息,则打印该i

lammps模拟“lost atoms”原子原因及解决方法
lammps模拟过程中,最怕的是模拟过程中出错,其中,比较常见的一个错误是“lost atoms”,也就是常说的原子丢失。 正常情况下,分子动力学模拟要保证原子数目保持不变。 nve、nvt、npt系综中的“n”就是原子数目,这些系综已经明确了模拟过程中要保证原子数目“n”不变,原子数目减少则会中止模拟,并给出“lost atoms”错误提示。 “lost atoms”错误出现的最主要的原因

lammps教程:读取文件提示“Did not assign all atoms correctly”的解决办法
大家好,我是小马老师。 lammps读取data文件时常见的一个错误:Did not assign all atoms correctly。 出现这种提示,基本就一个原因:建模方法不当造成部分原子位于box外面。 检验方法也比较简单,直接用ovito打开data文件,会发现有一些原子位于白色框的外面。 在lammps中,read_data命令只读取box内部的原子坐标,如果有原子位于box

“atoms are time integrated more than once”积分重复警告,常见三种原因及解决办法
大家好,我是小马老师。 本文介绍lammps in文件编写过程中一个常见的错误:重复积分。 分子动力学模拟的主要运算是原子的积分运算,通过求解积分方程得到原子的受力以及计算原子的速度,从而得出下一时刻原子的位置和状态。 原子的积分体现到代码中就是fix nve/nvt/npt等系综的设置,有些fix命令也自带积分运算,如fix move等命令。 原子不设置系综,就不会参与积分方程的求解,最明显的现
726. Number of Atoms
原题链接 思路 思路1 设置两个栈,一个放括号,另一个放Map,放一个左括号,就放一个Map,括号出栈时,把另一个栈头的map弹出,值加入到新的栈顶map。 public String countOfAtoms(String formula) {Deque<Character> stack1 = new ArrayDeque<>();Deque<Map<String, Integer>>

lammps案例 delete_atoms命令实现球壳结构建模
球壳结构在先进复合材料的发展中具有特殊的意义,因为它们可以有效地将不同的组分在纳米尺度上聚集在一起,这种结构的优点很大程度上依赖于核心和外壳的关键设计。本文将介绍一种球壳分子模型的lammps建立过程,本教程适合于新手,同时希望专业前辈提出不足。 如下图为本文建立的球壳结构模型。 内核为Al原子,外层包覆着一定厚度的Al原子。 (这里以Al原子为例,读者可自定义两种原子)

Leetcode 726. Number of Atoms
文章作者:Tyan 博客:noahsnail.com | CSDN | 简书 1. Description 2. Solution 解析:这道题还有优化的空间,这样写主要是逻辑清晰。1. 把元素(多个字母)、数字(多个数字字符)、左右括号拆分开;2. 计算元素的个数,如果元素后没有数字,则添加数字1作为元素个数;当碰到右括号时,查找其对应的左括号,并将其中的元素个数乘以括号后的数

CF 1425 - E. Excitation of Atoms
题目链接 题意: 给 n n n 个原子。每个原子有两种状态,静态和激发态。每个原子 i i i 从静态转换到激发态需要消耗 d [ i ] d[i] d[i] 的能量,而在激发态会贡献的 a [ i ] a[i] a[i] 的能量。初始的时候,序号从 1 − n 1-n 1−n 的原子是按照编号顺序连接在一起的,也就是原子 i i i 连接到原子 i + 1 i + 1 i
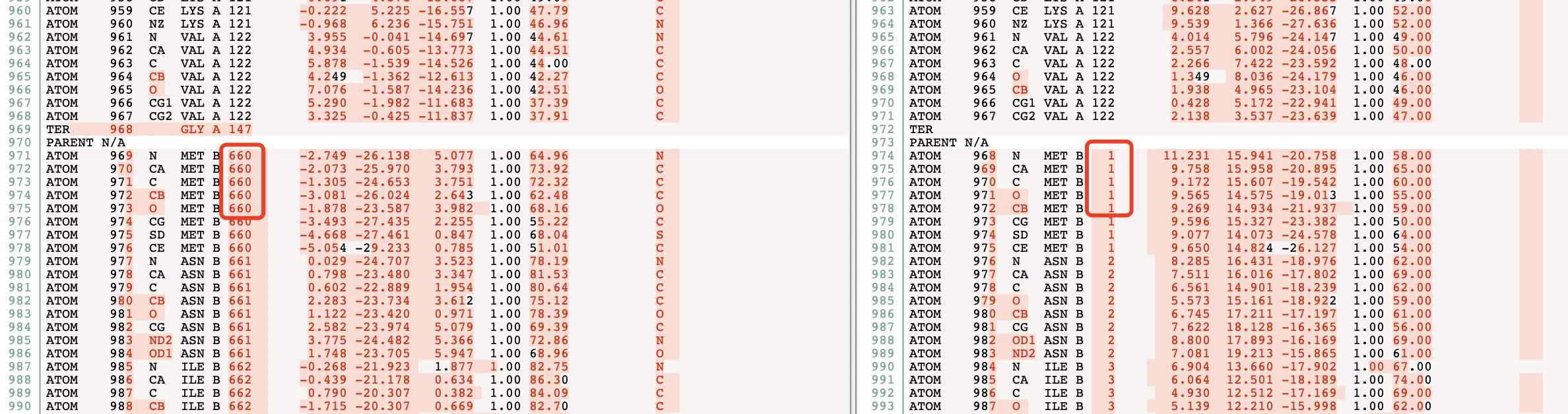
PSP - DockQ Zero number of equivalent atoms in native and model ligand (BugFix)
欢迎关注我的CSDN:https://spike.blog.csdn.net/ 本文地址:https://blog.csdn.net/caroline_wendy/article/details/129277522 ESMFold输出的PDB文件,不支持DockQ。 DockQ Bug: AssertionError: Zero number of equivalent atoms