本文主要是介绍蛋白质PDB文件解析+建图(biopython+DGL),希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
PDB文件解析
PDB文件设计得非常好,能够比较完整地记录实验测定数据
读懂蛋白质PDB文件-腾讯云开发者社区-腾讯云 (tencent.com)
科学网—PDB文件格式说明 - 李继存的博文 (sciencenet.cn)
从蛋白质结构来看,首先它会有多种不同的测定模型,然后每个模型中包含多条链,每条连上包含若干个残基,每个残基包含若干个原子
在biopython.PDB包中可以找到这些概念对应的模块:model、chain、residue、atom
Bio.PDB软件包 — Biopython 1.79 文档 (osgeo.cn)
首先用PDBParser读取文件,获得structure
struct = parser.get_structure(0,pdbfilepath)
model = struct.get_models().__next__()struct内部的一层结构是model,我们只取第一个model
然后就可以用循环遍历chains、residues、atoms
for chain in model.get_chains():for residue in chain.get_residues():for atom in residue.get_atoms():虽然也可以直接从structure中获取atom序列、residue序列、chain序列
但是这样就失去了一些从属关系
另外还可以通过查询的方式获得残基中的某个原子
例如使用residue['CA']就可以获取残基residue中名字为‘CA’的原子(氨基酸α碳原子)
所以我们只需要枚举残基,给每个残基的Cα原子进行编号就可以了
一般的PDB文件中都包含原子坐标数据,可以用它来作为这个氨基酸的一个特征
另外,为了获得序列,我们还需要把氨基酸的三字母缩写转化为一字母缩写,下面是一个转换矩阵。
three2one= {'VAL':'V', 'ILE':'I', 'LEU':'L', 'GLU':'E', 'GLN':'Q','ASP':'D', 'ASN':'N', 'HIS':'H', 'TRP':'W', 'PHE':'F', 'TYR':'Y', 'ARG':'R', 'LYS':'K', 'SER':'S', 'THR':'T', 'MET':'M', 'ALA':'A', 'GLY':'G', 'PRO':'P', 'CYS':'C'
}使用DGL对蛋白质建图
DGL库接口解析:dgl — DGL 2.1.0 documentation
建图的方法就很多了,得看个人的设计,有只根据Cα原子之间的距离进行建图的,有使用所有原子来建图的,也有用肽链原子+残基Cβ原子+二硫键建图的,只能说是五花八门,不过这些都只是多写几个if 的问题。
特征构造的方式就更多了
简单的,边不加特征,氨基酸作为点用独热向量作为特征
复杂的就多了去了,有加化学键长度的,有对化学键加入类别的,有加入残基直径的,还有加入原子数、分子量、氨基酸电性的,氨基酸特征向量还可以用一些序列比对的特征值,例如blosum,pam,cIndex of /blast/matrices (nih.gov) 总之想法很多,但真正有用的就那么几个。
DGL添加点:(第一个参数是点的个数,后面是一个dict,里面写这个点的特征向量tensor,但必须行数相同)
graph.add_nodes(1, {'pos': pos_feature, 'res': residue_feature})DGL添加边:(必须用两个tensor来表示两个端点的编号,后面的dict同样是边的特征向量)
值得注意的是,DGL只会添加单向边,所以无向边需要加两次
(ps:在网上看到许多写法都是graph=dgl.add_edges(graph,……),但graph.adde_edges()也是可以用的)
graph.add_edges(torch.tensor([atom_num-1]), torch.tensor([atom_num]), {'feat': Peptide_bond_feature})这里我使用的是,只根据肽键和Cα原子进行建图,以Cα原子代替氨基酸作为点,边为两个氨基酸之间是否存在肽键,点特征为氨基酸的blosum80特征和坐标特征,边特征为独热向量,方便以后加入其他类型的边。
import Bio.PDB.PDBParser
import os
import numpy
import torch
import dglparser = Bio.PDB.PDBParser()
# 连接Cα的肽键的边特征向量
Peptide_bond_feature = torch.tensor([1.0,0.0])
# 连接Cα的二硫键的边特征向量
S_S_bond_feature = torch.tensor([0.0,1.0])three2one= {'VAL':'V', 'ILE':'I', 'LEU':'L', 'GLU':'E', 'GLN':'Q','ASP':'D', 'ASN':'N', 'HIS':'H', 'TRP':'W', 'PHE':'F', 'TYR':'Y', 'ARG':'R', 'LYS':'K', 'SER':'S', 'THR':'T', 'MET':'M', 'ALA':'A', 'GLY':'G', 'PRO':'P', 'CYS':'C'
}
# 氨基酸的特征
AA_feature_blosum80 = {
'A' : torch.tensor([ 7, -3, -3, -3, -1, -2, -2, 0, -3, -3, -3, -1, -2, -4, -1, 2, 0, -5, -4, -1, -3, -2, -1]),
'R' : torch.tensor([-3, 9, -1, -3, -6, 1, -1, -4, 0, -5, -4, 3, -3, -5, -3, -2, -2, -5, -4, -4, -2, 0, -2]),
'N' : torch.tensor([-3, -1, 9, 2, -5, 0, -1, -1, 1, -6, -6, 0, -4, -6, -4, 1, 0, -7, -4, -5, 5, -1, -2]),
'D' : torch.tensor([-3, -3, 2, 10, -7, -1, 2, -3, -2, -7, -7, -2, -6, -6, -3, -1, -2, -8, -6, -6, 6, 1, -3]),
'C' : torch.tensor([-1, -6, -5, -7, 13, -5, -7, -6, -7, -2, -3, -6, -3, -4, -6, -2, -2, -5, -5, -2, -6, -7, -4]),
'Q' : torch.tensor([-2, 1, 0, -1, -5, 9, 3, -4, 1, -5, -4, 2, -1, -5, -3, -1, -1, -4, -3, -4, -1, 5, -2]),
'E' : torch.tensor([-2, -1, -1, 2, -7, 3, 8, -4, 0, -6, -6, 1, -4, -6, -2, -1, -2, -6, -5, -4, 1, 6, -2]),
'G' : torch.tensor([ 0, -4, -1, -3, -6, -4, -4, 9, -4, -7, -7, -3, -5, -6, -5, -1, -3, -6, -6, -6, -2, -4, -3]),
'H' : torch.tensor([-3, 0, 1, -2, -7, 1, 0, -4, 12, -6, -5, -1, -4, -2, -4, -2, -3, -4, 3, -5, -1, 0, -2]),
'I' : torch.tensor([-3, -5, -6, -7, -2, -5, -6, -7, -6, 7, 2, -5, 2, -1, -5, -4, -2, -5, -3, 4, -6, -6, -2]),
'L' : torch.tensor([-3, -4, -6, -7, -3, -4, -6, -7, -5, 2, 6, -4, 3, 0, -5, -4, -3, -4, -2, 1, -7, -5, -2]),
'K' : torch.tensor([-1, 3, 0, -2, -6, 2, 1, -3, -1, -5, -4, 8, -3, -5, -2, -1, -1, -6, -4, -4, -1, 1, -2]),
'M' : torch.tensor([-2, -3, -4, -6, -3, -1, -4, -5, -4, 2, 3, -3, 9, 0, -4, -3, -1, -3, -3, 1, -5, -3, -2]),
'F' : torch.tensor([-4, -5, -6, -6, -4, -5, -6, -6, -2, -1, 0, -5, 0, 10, -6, -4, -4, 0, 4, -2, -6, -6, -3]),
'P' : torch.tensor([-1, -3, -4, -3, -6, -3, -2, -5, -4, -5, -5, -2, -4, -6, 12, -2, -3, -7, -6, -4, -4, -2, -3]),
'S' : torch.tensor([ 2, -2, 1, -1, -2, -1, -1, -1, -2, -4, -4, -1, -3, -4, -2, 7, 2, -6, -3, -3, 0, -1, -1]),
'T' : torch.tensor([ 0, -2, 0, -2, -2, -1, -2, -3, -3, -2, -3, -1, -1, -4, -3, 2, 8, -5, -3, 0, -1, -2, -1]),
'W' : torch.tensor([-5, -5, -7, -8, -5, -4, -6, -6, -4, -5, -4, -6, -3, 0, -7, -6, -5, 16, 3, -5, -8, -5, -5]),
'Y' : torch.tensor([-4, -4, -4, -6, -5, -3, -5, -6, 3, -3, -2, -4, -3, 4, -6, -3, -3, 3, 11, -3, -5, -4, -3]),
'V' : torch.tensor([-1, -4, -5, -6, -2, -4, -4, -6, -5, 4, 1, -4, 1, -2, -4, -3, 0, -5, -3, 7, -6, -4, -2]),
'B' : torch.tensor([-3, -2, 5, 6, -6, -1, 1, -2, -1, -6, -7, -1, -5, -6, -4, 0, -1, -8, -5, -6, 6, 0, -3]),
'Z' : torch.tensor([-2, 0, -1, 1, -7, 5, 6, -4, 0, -6, -5, 1, -3, -6, -2, -1, -2, -5, -4, -4, 0, 6, -1]),
'X' : torch.tensor([-1, -2, -2, -3, -4, -2, -2, -3, -2, -2, -2, -2, -2, -3, -3, -1, -1, -5, -3, -2, -3, -1, -2]),
}def get_seq_graph_info(pdbfilepath):struct = parser.get_structure(0,pdbfilepath)model = struct.get_models().__next__()seq = []graph = dgl.DGLGraph()residue_num = 0for chain in model.get_chains():residues = chain.get_residues()# print(chain)chain_start_flag = 1for residue in residues:res_name = three2one[residue.get_resname()]seq.append(res_name)pos_feature = torch.from_numpy(residue['CA'].get_coord()).unsqueeze(0)residue_feature = AA_feature_blosum80[res_name].unsqueeze(0)graph.add_nodes(1, {'pos': pos_feature, 'res': residue_feature})if chain_start_flag == 0 :graph.add_edges(torch.tensor([residue_num-1]), torch.tensor([residue_num]), {'feat': Peptide_bond_feature})graph.add_edges(torch.tensor([residue_num]), torch.tensor([residue_num-1]), {'feat': Peptide_bond_feature})residue_num += 1chain_start_flag = 0return seq,graphpdb_data_dir = 'D:\PVT\data'
file_list = [os.path.join(pdb_data_dir, file) for file in os.listdir(pdb_data_dir)]
for file in file_list:if file.endswith('.pdb'):print("processing "+file)seq,graph = get_seq_graph_info(file)print(seq)print(graph)print("finished!")运行结果:
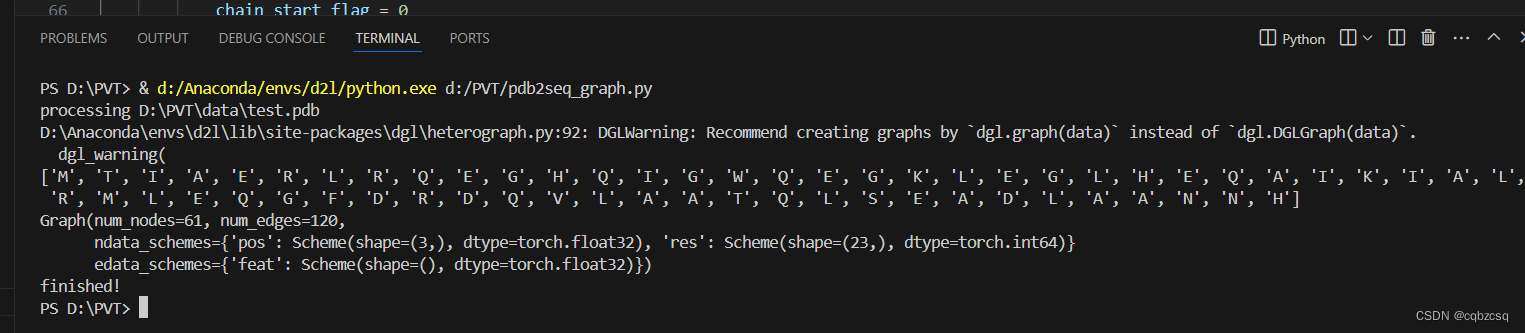
点数61,边数120
可以看出这个蛋白质只是一条肽链
这样我们就从PDB文件中得到了 氨基酸序列和蛋白质的图
接下来就可以愉快地使用GNN之类的东西提取特征啦
这篇关于蛋白质PDB文件解析+建图(biopython+DGL)的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!





