本文主要是介绍【Schrödinger薛定谔软件使用实战】- 4lyw蛋白实战,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
文章目录
- 软件选择
- 1 pretein preparation
- 1.1 import and process注意
- 1.1.1 preprocess可能遇到的问题
- 1.2 review and modify
- 1.3 refine
- 1.3.1 optimize优化氢键网络
- 1.3.2 minimize 氢原子会进行能量最小化
- 2 ligand prepare
- 3 生成对接盒子-receptor grid generation
- 3.1 receptor-确定受体
- 3.1.1 define receptor
- 3.1.2 van der waals radius scaling
- 3.2 site 设置对接盒子大小
- 3.2.1 advanced settings
- 3.3 contraints-设置约束条件 (例如受体配体之间的氢键)
- 3.3.1 positional/NOE(保持风格,要出现在特定范围内
- 3.3.2 H-bond/Metal 黄线-氢键作用 或配体和金属原子相互作用(并不要求方向)
- 3.3.3 Metal Coordination 金属配位作用-可以调节方向
- 3.4 Rotatable Groups-规定哪些羟基等是可以旋转的
- 3.5 Excluded Volumes
- 4 ligand grid
- 4.1 ligands选择已经准备好的配体
- 4.2 setting
- 4.2.1 设置对接准确度
- 4.2.2 ligand sampling 配体选择采样
- 4.2.3 选择采样偏向,三种选项
- 4.3 contraints选择对3的限制条件的选择
- 4.4 output 输出格式选择
- 注意事项 PDB格式
软件选择
采用的是Maestro 12.8版本
1 pretein preparation
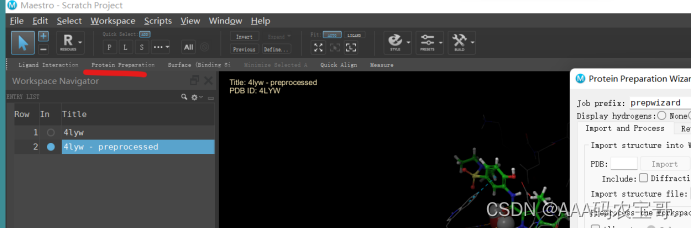
1.1 import and process注意
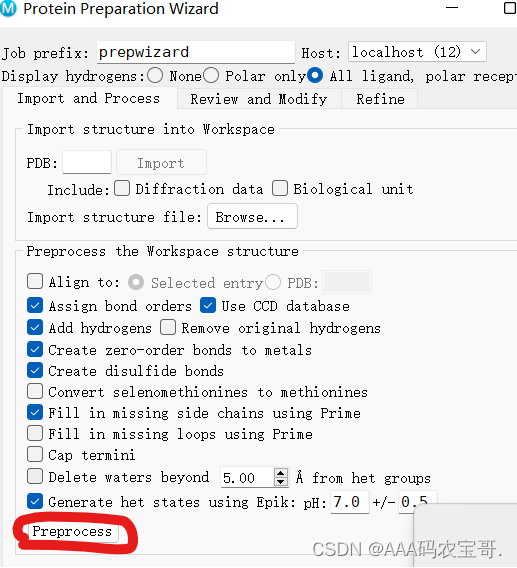
- 1、有loop的时候,如果口袋在loop区域,pretein preparation一定要加上fill in loops
- 2、准备蛋白时,注意对保守水分子的选择
- 3、对于PH的选择,注意应当去质子还是被质子化
!!!!!点击preprocess
1.1.1 preprocess可能遇到的问题
- alternates postitions:对于alternates postitions的残基选择,蛋白具有不同构象,注意选择,如果离得远,随便选,离得近就要认真考虑
- overlapping atoms:对于氢原子会进行能量最小化的处理
1.2 review and modify
若小分子有很多带电状态,进行选择
1.3 refine
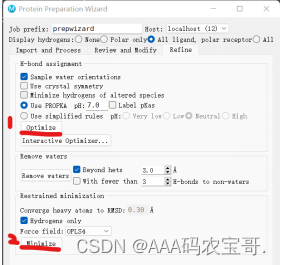
1.3.1 optimize优化氢键网络
选用默认选项,entry list增加hbond-opt
!!!!!点击optimize
1.3.2 minimize 氢原子会进行能量最小化
如果存在里的近的,橙黄线,尝试点击minimize消掉
!!!!!点击minimize
选用默认选项,entry list增加minimized
2 ligand prepare
import structure

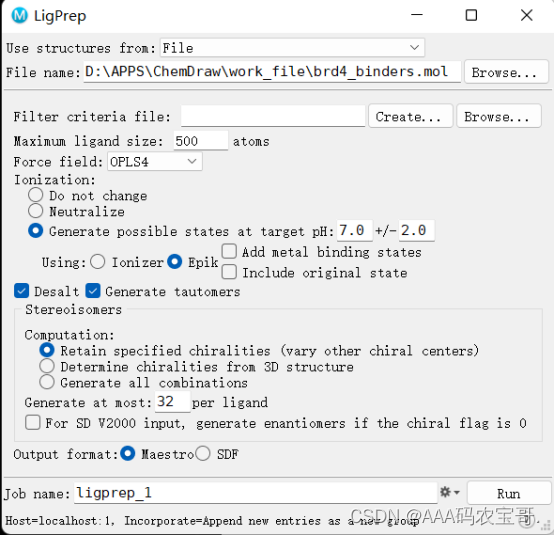
设置筛选条件
-
-general attributes
Molecular_weight 分子量 <=150 and >=500
Num_chiral_centes 手性分子 -
-functional group counts
Aldehydes 醛基数目》=1
PH严重影响配体的质子化状态


-
若对接有金属离子,则加上add metal binding states
如果配体是2D结构computation选择第一个
如果是3D选择第二个

3 生成对接盒子-receptor grid generation
3.1 receptor-确定受体
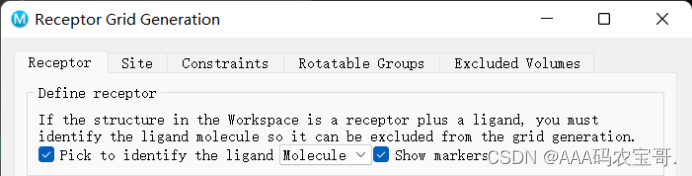
3.1.1 define receptor
点击show markers 标注小分子,使其不会当作为受体的一部分
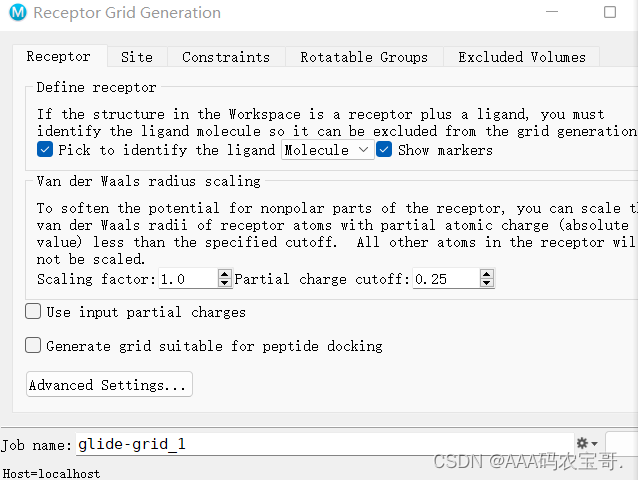
3.1.2 van der waals radius scaling
范德华半径的缩放–Glide:小分子是柔性的,受体是刚性的
对接分为刚性对接、柔性对接、半柔性对接
3.2 site 设置对接盒子大小
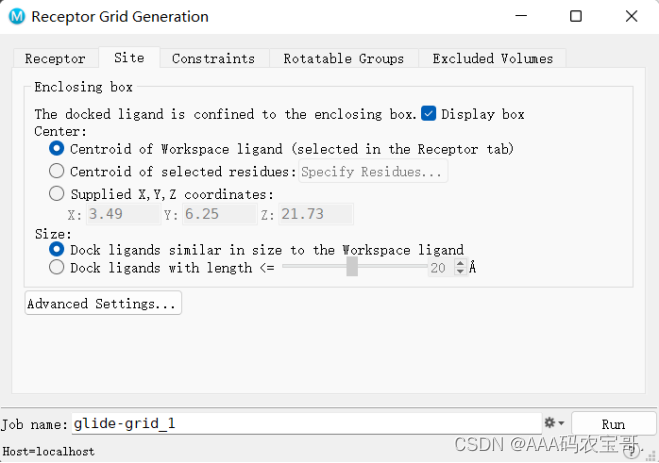
3.2.1 advanced settings
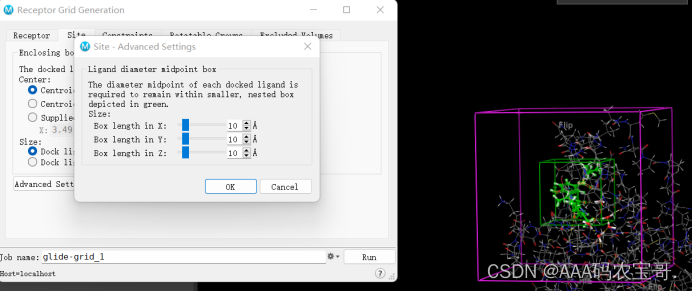
紫色盒子更大()
绿色盒子设置配体中心(对接时小分子中心点可以去到的位置)
若work_space只有受体,没有配体的话是没有办法通过点击配体来选择配体并生成紫色盒子,这样需要通过选择活性位点的氨基酸残基来定义对接盒子
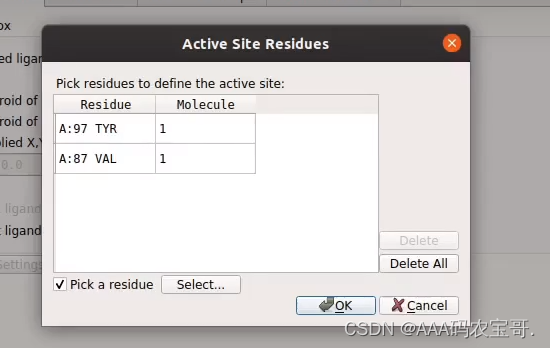
通过select来选择氨基酸残基
3.3 contraints-设置约束条件 (例如受体配体之间的氢键)
来在早期去除掉不符合的配体模式
3.3.1 positional/NOE(保持风格,要出现在特定范围内
要求配体也在该区域有类似的组成,重视位置
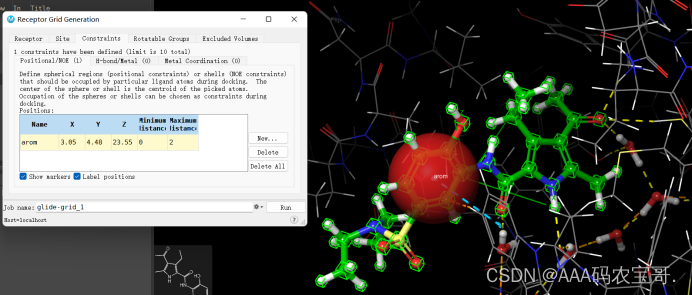
NOE-针对氨基酸残基的距离,配体小分子出现在哪里等
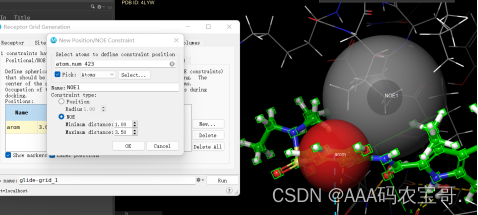
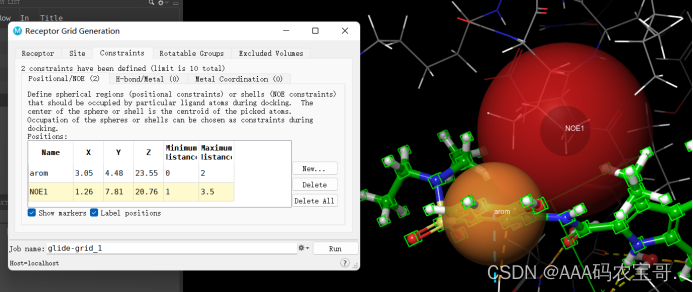
3.3.2 H-bond/Metal 黄线-氢键作用 或配体和金属原子相互作用(并不要求方向)
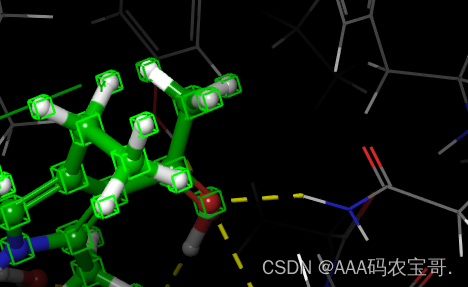
配体羰基与天冬酰胺140有氢键作用
哪些氢键特别重要,则需要在H-bond/Metal中标出-选择天冬酰胺140的氢原子
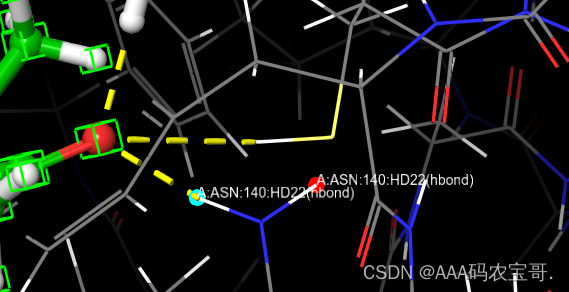
gilde自动标注对称原子
点击use symmetry去除距离较远氢原子
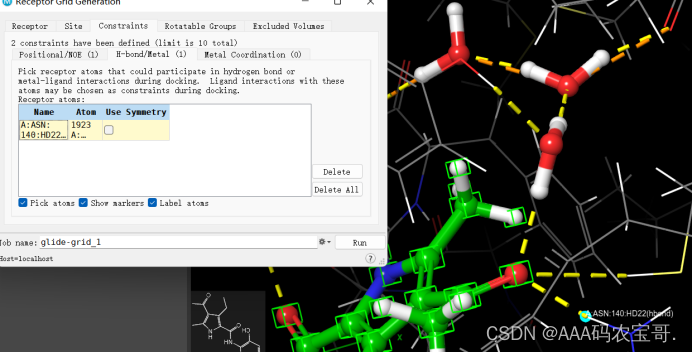
3.3.3 Metal Coordination 金属配位作用-可以调节方向
3.4 Rotatable Groups-规定哪些羟基等是可以旋转的
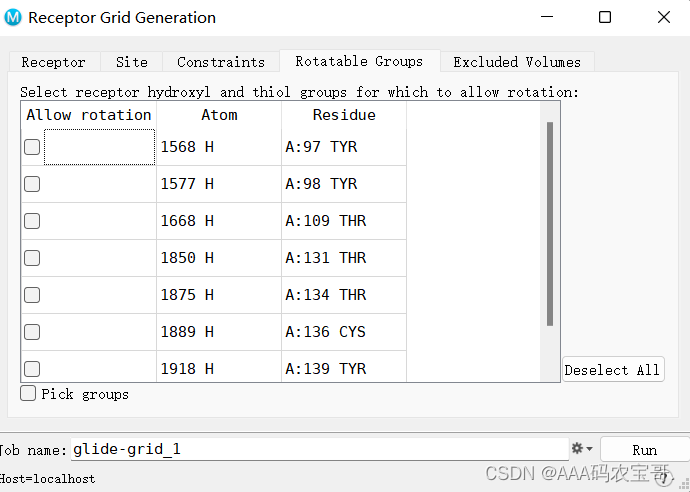
pick groups -选择残基
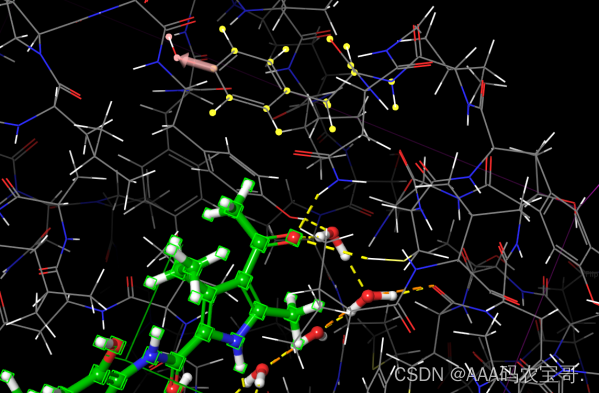
选择酪氨酸,允许酪氨酸进行旋转
旋转在grid时是不可清除的
3.5 Excluded Volumes
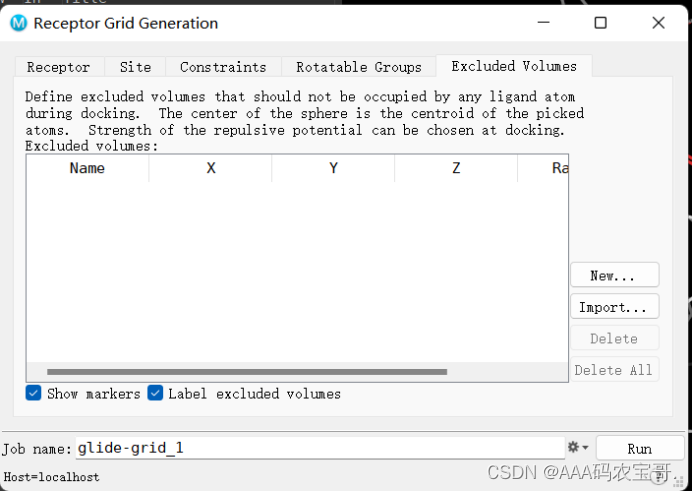
排除口袋位置的小分子
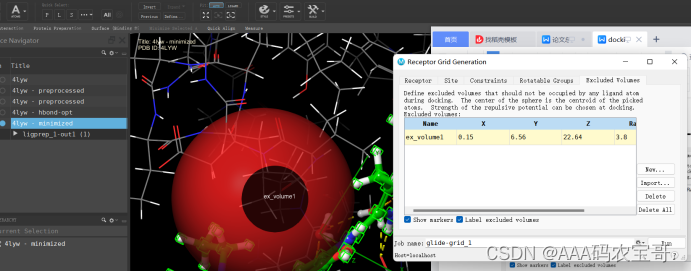
对接时,任何小分子的原子不能出现在红色范围内
对接口袋规定内部里面的物理化学特性
4 ligand grid
选择ligand docking
display receptor

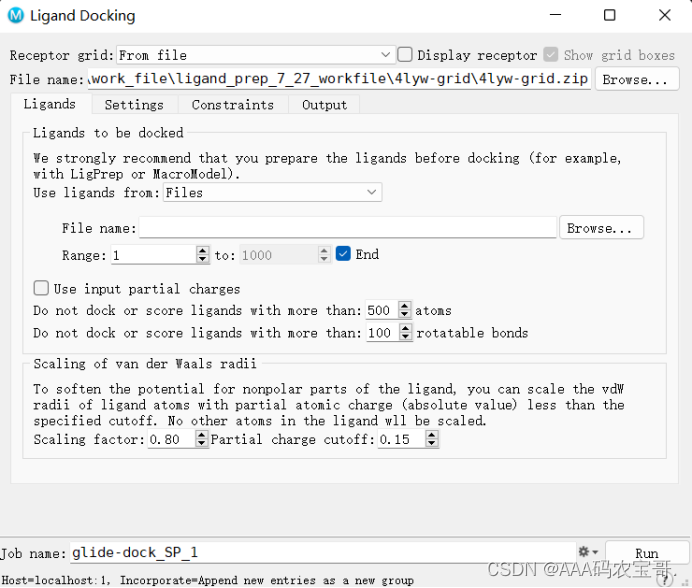
4.1 ligands选择已经准备好的配体

是否使用输入文件自带的局部电荷
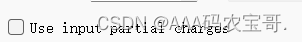
默认不会对接分子量超过500 旋转超过100的,按需调整
调节配体的非极性范德华力的缩放

4.2 setting
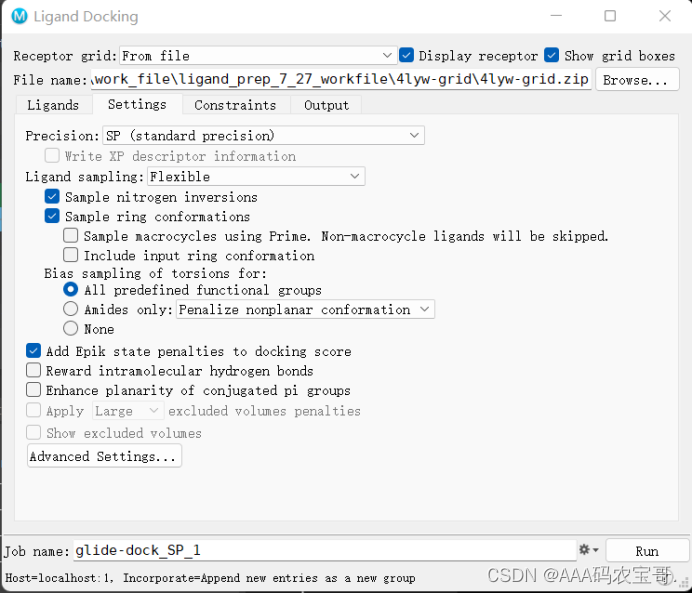
4.2.1 设置对接准确度
- precision
HTVS 通量虚拟筛选,快速筛选小分子,更为严格,不可以score inplace(通常打分和对接是两个软件在做)
XP 与SP类似,但对形状互补更严格,计算量更大,打分更严格
建议:
筛选大数据库时,先用sp,再按照打分排序,取前10%~30%,用xp重新对接,可以勾选
若超大虚拟筛选,建议采用另一模块
4.2.2 ligand sampling 配体选择采样
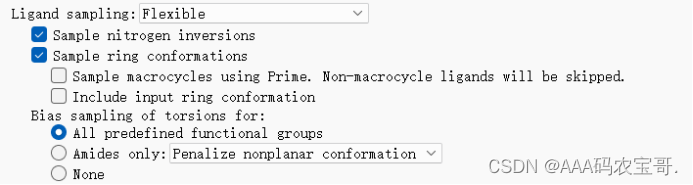
选择配体柔性,允许inverse,并且考虑环采样
4.2.3 选择采样偏向,三种选项
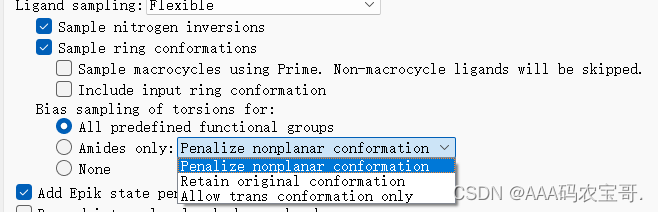
第一个对资源文件的选择进行扭转偏斜
第二个是对酰胺的C和N的选择:1、惩罚非平面的构象(允许酰胺变形,非顺式等)2、保留酰胺键的输入原始构象3、只允许反式20°角的偏斜
4.3 contraints选择对3的限制条件的选择
4.4 output 输出格式选择
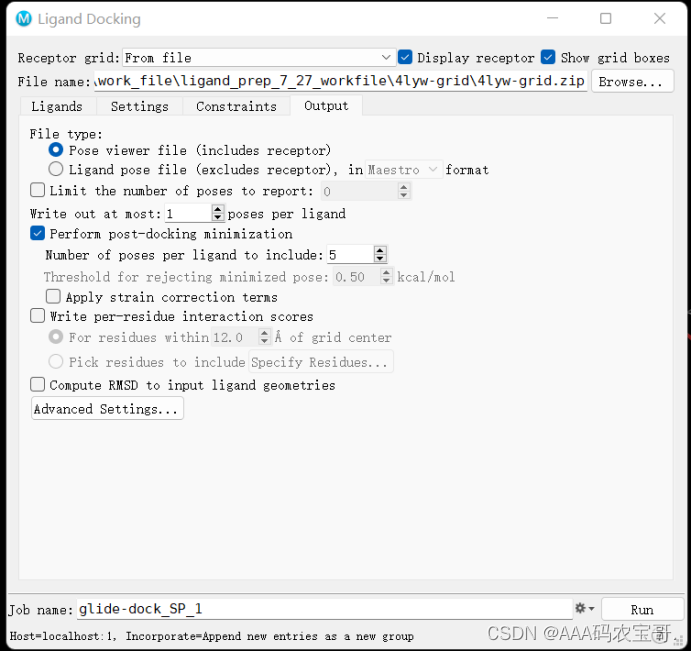
若想查看每个氨基酸基团对于构象的打分贡献的话可以勾选
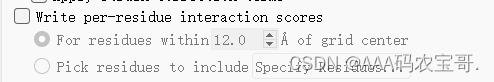
若进行虚拟筛选很大时,点击run旁边齿轮,选择Do not incorporate
剩下的点击run即可
注意事项 PDB格式
Pdb
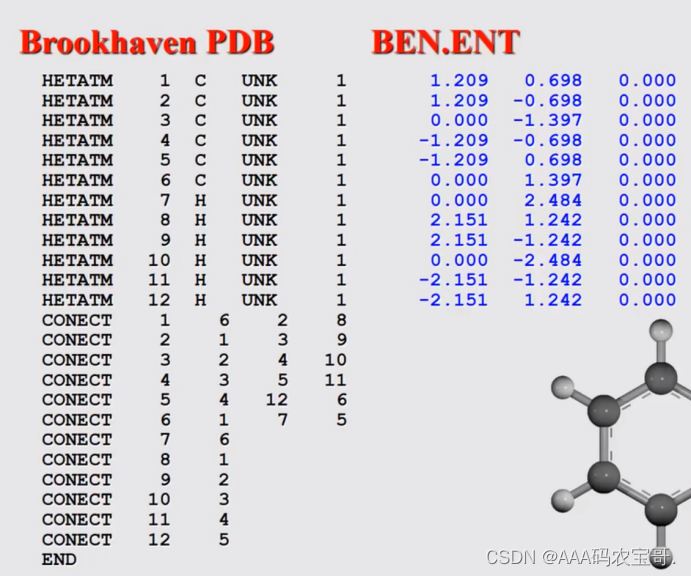
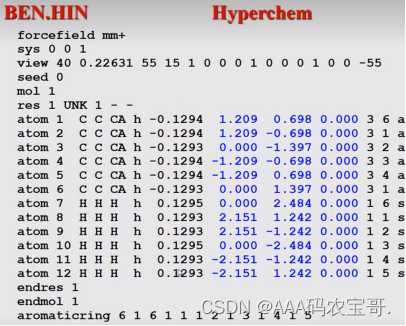
Hyperchem mm+力场中 -0.1294是电荷数
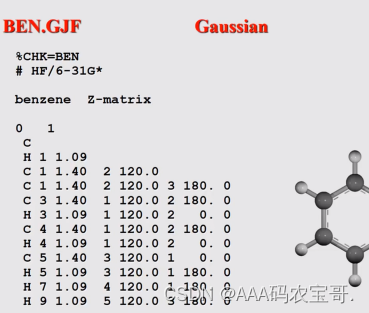
Gaussian z-矩阵,采用分子内坐标 键角 二面角
这篇关于【Schrödinger薛定谔软件使用实战】- 4lyw蛋白实战的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!






