本文主要是介绍BERT一个蛋白质-季军-英特尔创新大师杯冷冻电镜蛋白质结构建模大赛-paipai,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
关联比赛: “创新大师杯”冷冻电镜蛋白质结构建模大赛
解决方案
团队介绍
paipai队、取自 PAIN + AI,核心成员如我本人IvanaXu(IvanaXu · GitHub),从事于金融科技业,面向银行信用贷款的风控、运营场景。但我们团队先后打过很多比赛,其中跨领域居多,如天文、海洋,也非常有幸参加本次蛋白质结构建模大赛。我们将延续“他山之石,可以攻玉”的基本思想,这也将在后续方案中体现。
主要方案
01 赛题解析

金融场景下其实就是在追求精准定位问题,并以最小成本换取更大收益。同样的,从现有解析蛋白质结构的主流方法来看,能覆盖较多场景(如生物大分子、静动两态、蛋白质折叠等)且精度较高,但为此需要付出大量人力成本,甚至是在专业软件辅助下才能完成的人工操作。另外,由DeepMind研发的AlphaFold2却能高效几乎0人工输出蛋白质结构,当然这个代价是覆盖场景有限(如生物大分子易错)、局部精度高(需要强依赖于已知蛋白质结构/PDB)。据此我们提出的解决方案是,拥抱类似于AlphaFold2的AI预测技术但充分结合传统方法,或许是最大收益方案。
即复杂生物应用场景下,求一种高效、准确的蛋白质结构解析方法。
所以我们将解决方案聚焦到以下两点:
(1)仅从氨基酸序列出发的AI预测技术为主,通过已知蛋白质结构去做一些自衍生处理,尝试去摆脱对已知蛋白质结构高度依赖;
(2)当遇到已知蛋白质结构甚至是自衍生蛋白质结构都不能很好识别时,再来考虑结合冷冻电镜数据,作为预测结果的辅助性修正,毕竟获取冷冻电镜数据就已经投入了一定的成本;
02 算法设计

如示例2145这个氨基酸序列,我们看到前4位的MSSK、就是一个甲硫氨酸+两个丝氨酸+一个赖氨酸的序列,但我们将它转为以下文本处理:
第一步,将氨基酸序列-蛋白质结构作为一组X-Y对,即使用氨基酸序列预测蛋白质结构,并计划进行自衍生处理1:将衍生后的氨基酸序列-蛋白质结构对进行随机混淆打乱,这一步复赛阶段未实现,但后续我们做了一些尝试;
第二步,将氨基酸序列转化为文本序列,在这里会做自衍生处理2:将它切分为不同长度的文本序列,如左侧V2算法的第一步,将MSSKS每隔4个氨基酸就切分为MSSK、SSKS等等,同样的还会按照5、6不等的长度进行切分,最终使用4、5、6、8、10、12、15、18,分别代表了4、5、6长度段的1倍、2倍、3倍。从生物意义上就代表了一些短氨基酸序列。这种切分方法的优点是可以是因为把其充分序列化,那么蛋白质中的对称重复架构会在序列中特别突出,有点像121213这组数字,我们听一遍后,在脑海中对12、121、21这样的组合印象就很深刻;
第三步,文本处理:尝试提取序列之间的潜在关系,事实上可考虑引入谷歌提出的基于Transformers的双向编码语言模型BERT来解决,但BERT很重,考虑上述成本效益,在此简化应用TF-IDF来处理;
第四步,将处理后序列信息输入至预测模型,输出最接近蛋白质结构;
为进一步比较自衍生处理1、自衍生处理2、以及不同文本处理算法之间的差异,我们先后设计了V1、V2、V3三个算法版本:
- V1,无自衍生处理1、无自衍生处理2,即MSSK序列会当作M、S、S、K四个单词组成的一句话,直接使用BM25文本模型进行预测;
- V2,在V1基础上增加自衍生处理2,即MSSK序列会切分为如图MSSK、SSKS、等等单词组成的一句更长的话;
可以通过比较V1、V2来体现自衍生处理2带来的预测增益。

- V3,会更充分考虑实际蛋白质结构预测中会遇到的问题,并根据本次比赛提供的E-HPC阿里云弹性高性能计算平台改进算法底层一些计算设计,如图:
(1)在高性能计算平台上应用CPU多核并发,能同时计算多个蛋白质,并将氨基酸序列计算转入稀疏矩阵存储 + TF-IDF限制某些高频率出现的简单序列,减少大量空值存储和无效计算,充分压缩氨基酸序列结果,避免内存错误;
(2)在V2的基础上调整BM25文本模型至TF-IDF+LR;
这就是复赛阶段的解决方案。
(3)另外,复赛后我们还在此基础上设计如左虚框部分的自衍生处理1,对算法拓展性做了一部分探索。
引入BERT中MLM/Masked Language Model即遮蔽语言模型的概念,其灵感来自于我们学英语很熟悉的完形填空,如我们通过线上会议XX了答辩,就会填写“参加”这个词。
这种概念其实类似于自衍生处理2,本质上是为了充分提取氨基酸序列的潜在关系,如前面提出的不同长度切分就为了获取一定的上下文信息,即氨基酸序列中S丝氨酸前后的氨基酸信息,但这种方式也仍然是单向或一个正向+一个反向这种假双向。反之MLM能更充分的解决这个问题,因为这个时候模型学到不是一个向量,而是**“一种学习能力”**。
TODO
- 所以在AAAI2020的工作中,已经有论文提出K-BERT即知识图谱+BERT的概念,使用类似MLM的方式尝试进行知识发现。
同样的,可以使用这个概念来“知识发现”一些未知的蛋白质结构,进而打破上述对已知蛋白质结构的高度依赖。
以下我们来对照AlphaFold2,对比一下上述算法:
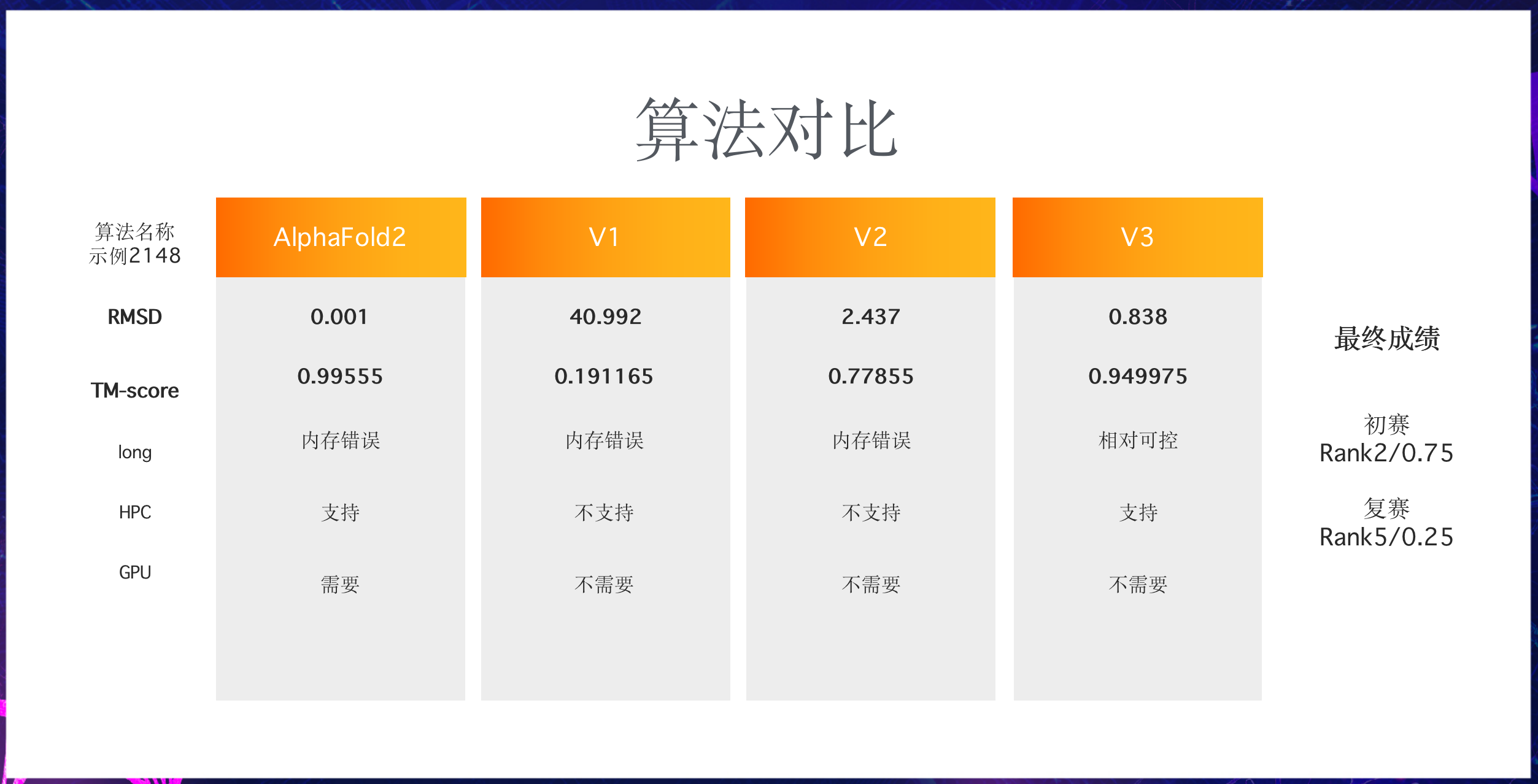
以2148为例,我们也尝试使用了AlphaFold2:
- 通过前两栏的RMSD、TM-score来看:
无疑是AlphaFold2最强,但对比V2、V1可以看到,在做了自衍生处理2即序列切分后,TM-score自0.19提升到了0.77,V2算法也就是初赛核心方案。另外,改进后的V3算法从个例来看相对接近于AlphaFold2。
- 另外,由于我们在调研阶段从PDB中发现了远比训练数据更大的蛋白质,所以这里的long评估项即长氨基酸序列的兼容,AlphaFold2/V1/V2都不同程度地出现在长氨基酸序列计算时内存错误的情况。前面介绍过,V3在做了一些相关设计所以相对可控。且能在不需要GPU的情况下进一步兼容HPC高性能计算,尽管AlphaFold2也提出支持HPC,对此我们未深入体验。
接下来我们看一下可视化结果对比:
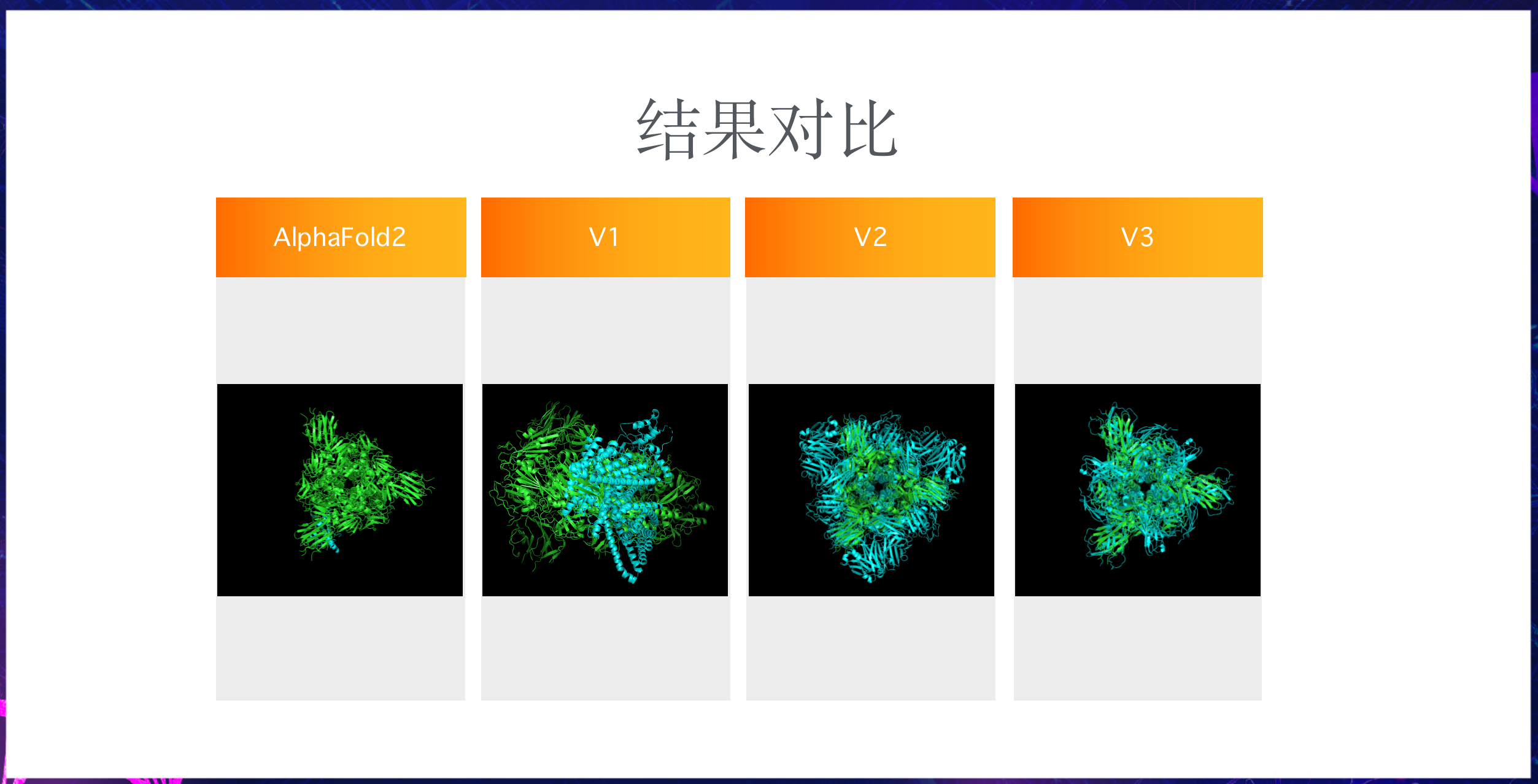
AlphaFold2已经接近严丝合缝,V1差异还比较大,但V2、V3已经有了相似的蛋白质结构雏形,未来可能可以进一步优化。
03 总结

1、直接转换为文本问题进行解决
事实上我们也发现其实这与Alphafold2的序列处理有类似的地方,但不同AI场景下2D 和3D transformers的发展,可能还可以进一步借鉴。但氨基酸序列的潜在关系我们还是交由更为简单的文本处理算法(TF-IDF)进行提取,这里考虑降低模型复杂性、提高应用效率。
如我们还没有提到的冷冻电镜图片处理,CVPR2022的工作中已经提出了Point-BERT基于掩码建模的3D点云自注意力模型,实现将类似于冷冻电镜数据这种3D结构表达为一个“词汇”集合。跟我们上述将氨基酸序列转化为文本处理的逻辑类似,我们的解决方案也提出了类似的思想,先用氨基酸序列进行文本预测,再对预测不佳的部分根据冷冻电镜数据也进行文本预测,作为辅助性修正,但这部分还没有来得及完成。
2、避免高度依赖已知蛋白质结构
为此前面我们设计了自衍生处理1、自衍生处理2,并通过V1/V2比较证明了自衍生处理2的有效性。也正是引入自衍生处理1,通过V2/V3比较看到了突破这种蛋白质结构依赖带来的额外效益。
相关参考
概念
-
清华大学结构生物学高精尖创新中心
https://ww.icsb.tsinghua.edu.cn
-
Alphafold2
alphafold.ebi.ac.uk
-
一键构建云上高可用蛋白质结构预测平台(一)
基于Alphafold2一键构建云上高可用蛋白质结构预测平台 | 亚马逊AWS官方博客
-
PDB,全称Protein Data Bank,是目前最主要的收集蛋白质三维结构的数据库
文章
- 当AI“进击”蛋白质结构预测
- 颜宁等点评:AI精准预测蛋白质结构,结构生物学何去何从?
- 颜宁点评AlphaFold2 + 外行买家秀:蛋白结构预测神器初体验
论文
- CN104951669A - 一种用于蛋白质结构预测的距离谱构建方法 - Google Patents
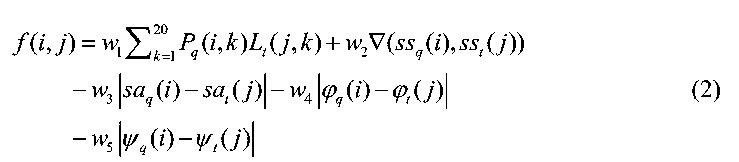
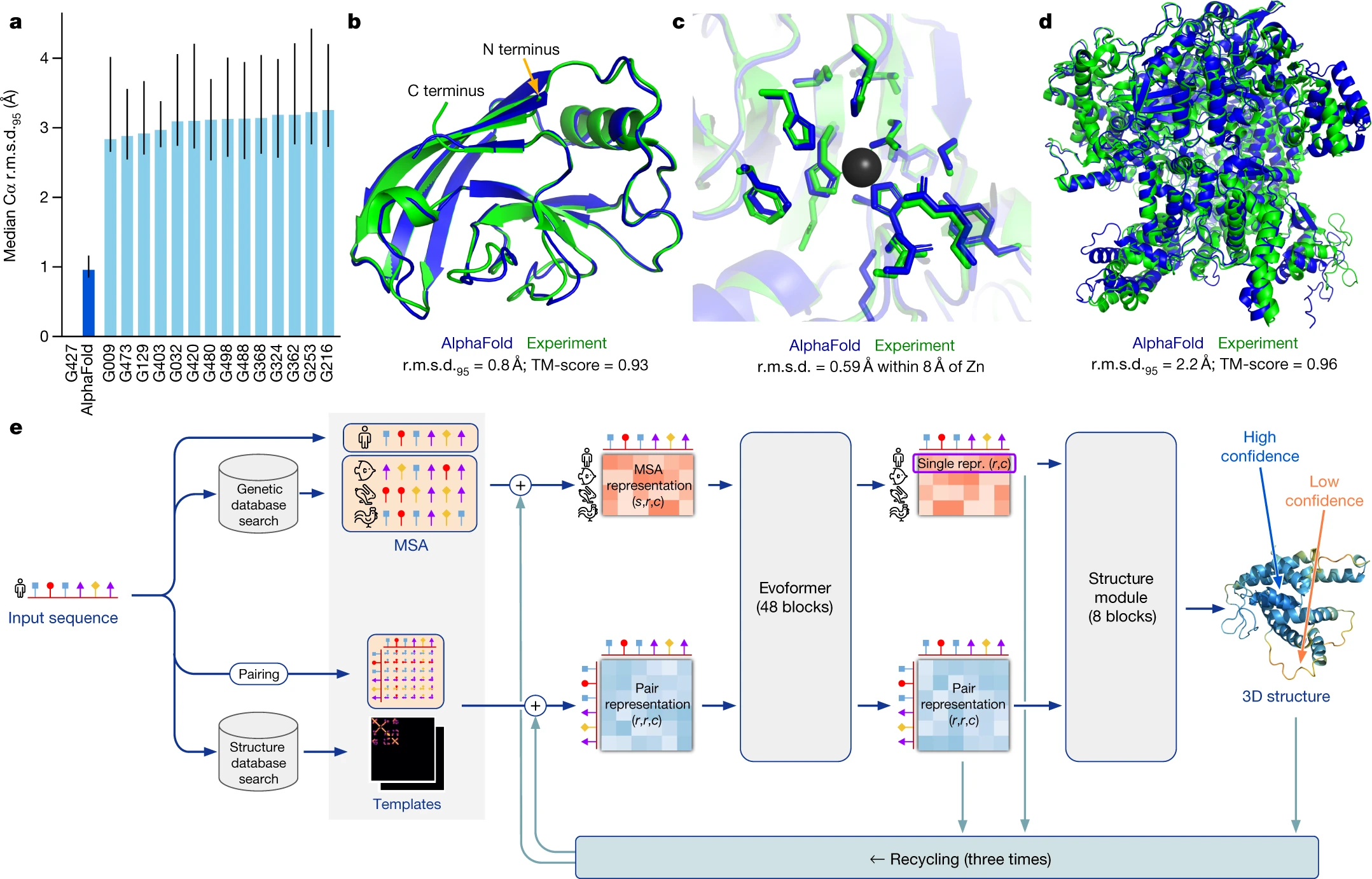
- Highly accurate protein structure prediction with AlphaFold
详见https://github.com/IvanaXu/TianChiProj/tree/master/ProteinStructureModeling
BERT一个蛋白质-季军-英特尔“创新大师杯”冷冻电镜蛋白质结构建模大赛-IvanaXu
查看更多内容,欢迎访问天池技术圈官方地址:BERT一个蛋白质-季军-英特尔创新大师杯冷冻电镜蛋白质结构建模大赛-paipai_天池技术圈-阿里云天池
这篇关于BERT一个蛋白质-季军-英特尔创新大师杯冷冻电镜蛋白质结构建模大赛-paipai的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!







