本文主要是介绍scDEA一键汇总12种单细胞差异分析方法 DESeq2、edgeR、MAST、monocle、scDD、Wilcoxon,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
问题来源
单细胞可以做差异分析,但是究竟选择哪种差异分析方法最靠谱呢?
解决办法
于是我去检索文献,是否有相关研究呢?
https://academic.oup.com/bib/article/23/1/bbab402/6375516文章指出,现有的差异分析方法(包括用于 bulk RNA-seq 和 scRNA-seq 的方法),它们都对样本量和批次效应敏感。
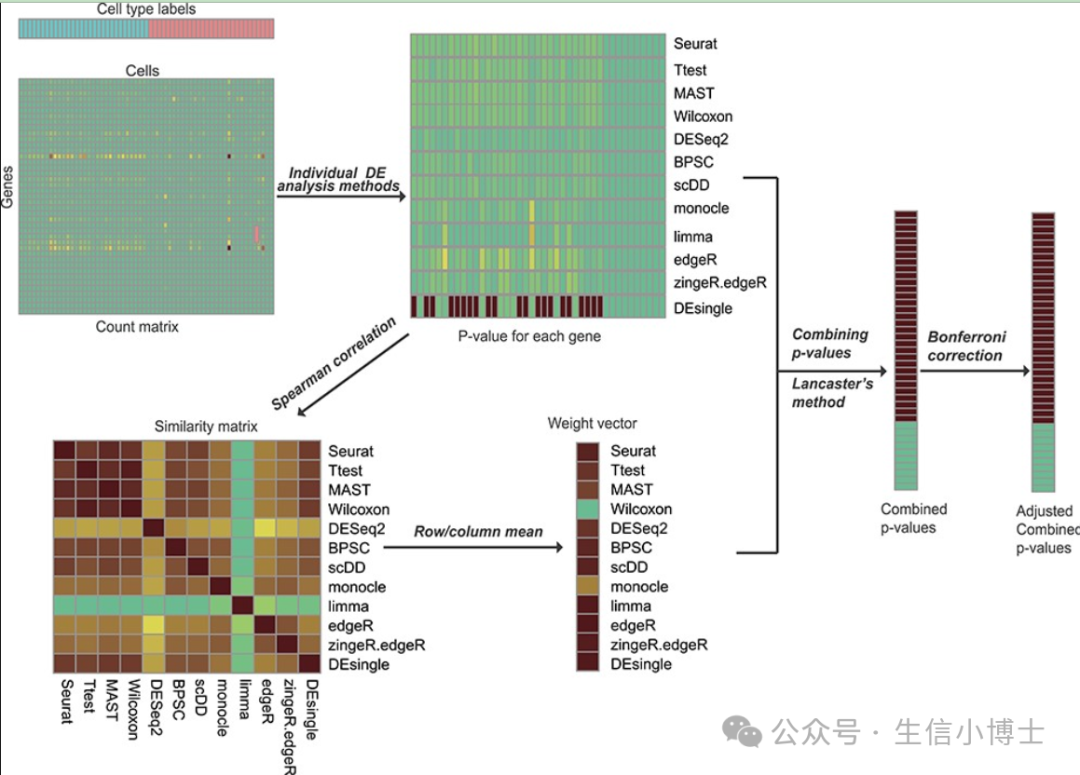
于是本研究没有执着于寻找现有技术中的最佳方法,也没有执着于开发一种新的高精度和稳健的方法,而是借鉴了现有算法的优势,开发了一种基于集成学习的 scRNA-seq 数据差异表达分析方法,名为 scDEA。scDEA 利用 Lancaster 组合概率测试方法,整合了来自 12 种独立方法( 既涵盖 bulk RNA-seq 也涵盖 scRNA-seq 数据) 的分析结果,从而得出一个准确且稳健的共识结果。
scDEA 在控制 I 型错误率、富集特定生物功能的差异表达基因检测、以及对不同样本量和不同实验批次的数据集识别出的一致性差异表达基因方面,都优于单个的差异表达分析方法。为了提供用户友好的工具,作者还开发了一个 R 软件包来实现集成分析过程 (https://github.com/Zhangxf-ccnu/scDEA)。此外,还开发了一个 Shiny 应用,以方便用户更轻松地实施和可视化分析结果 (https://github.com/Zhangxf-ccnu/scDEA-shiny)。
.scDEA 的工作原理
scDEA 主要包含以下四个步骤:
对原始计数矩阵进行归一化,以满足各个差异表达分析方法的要求
运行 12 种差异表达分析方法,并为每个基因生成 p 值
整合各个方法生成的 p 值,以获得一致的结果
对整合后的 p 值进行调整,以解决多重检验问题
注意,对于scDEA,我们需要使用counts矩阵哦:
由于一些差异表达分析框架只接受原始计数矩阵,而其他方法则需要归一化表达矩阵作为输入,为了给所有 12 种独立的差异表达分析方法构建统一的输入,scDEA 首先将原始计数矩阵转换为每百万 (CPM) 比对 reads,然后将原始计数矩阵、CPM 归一化表达矩阵和细胞类型标签一起集成,并使用 R 软件包 SingleCellExperiment [20] 保存为一个 SingleCellExperiment 对象。
scDEA R包安装
.scDEA 软件包的依赖
scDEA 软件包依赖以下 R 软件包:BPSC、DEsingle、DESeq2、edgeR、MAST、monocle、scDD、limma、Seurat、zingeR、SingleCellExperiment、scater 和 aggregation。安装 scDEA 时,它可以加载大部分的依赖包。但是,如果依赖项没有正确安装,则需要您自己手动安装。
.Windows 系统上的注意事项
根据经验,scDEA 在 Windows 系统上无法自动安装 BPSC、DEsingle 和 zingeR 软件包,但是在 Ubuntu 16.04 系统上可以加载所有依赖包。因此,如果您在 Windows 系统上使用 scDEA,建议 首先先安装 BPSC、DEsingle 和 zingeR 软件包。
devtools::install_github("nghiavtr/BPSC")BiocManager::install("DEsingle")devtools::install_github("statOmics/zingeR")devtools::install_github("Zhangxf-ccnu/scDEA")
成功安装:
但是,官方教程太简陋了

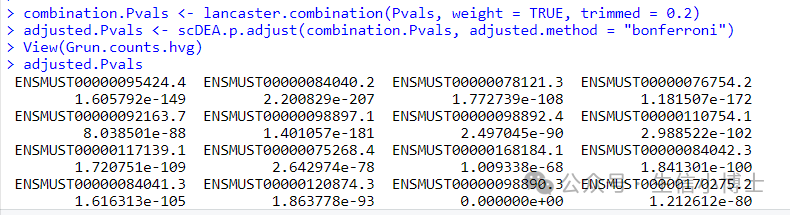
data("Grun.counts.hvg")data("Grun.group.information")Pvals <- scDEA_individual_methods(raw.count = Grun.counts.hvg, cell.label = Grun.group.information)combination.Pvals <- lancaster.combination(Pvals, weight = TRUE, trimmed = 0.2)adjusted.Pvals <- scDEA.p.adjust(combination.Pvals, adjusted.method = "bonferroni")
还好,示例数据是可以运行的,激动!

最后每个基因都能得到一个p值,但是奇怪的是,作者并没有给出差异基因的表格(我已经问了作者,静候回复)。但是我们可以退而求其次,把findmarker得到的结果和这个p值取交集,进而得到差异基因。
作者说scDEA这种方法可以减少一类错误,减少细胞数目的影响,减少批次效应的影响......
.下面,我使用自己的pbmc数据测试一下.
如果你对示例数据不熟悉,可以参考我之前的推文与直播
1 20231126单细胞直播一理解pbmc处理流程与常用R技巧
2 单细胞直播一理解seurat数据结构与pbmc处理流程
3 单细胞数据pbmc数据下载,标准处理流程获取seurat对象
scDEA 主要包含以下四个步骤:
对原始计数矩阵进行归一化,以满足各个差异表达分析方法的要求
运行 12 种差异表达分析方法,并为每个基因生成 p 值
整合各个方法生成的 p 值,以获得一致的结果
对整合后的 p 值进行调整,以解决多重检验问题
library(scDEA)load("~/gzh/pbmc3k_final_v4.rds")head(pbmc@meta.data)as.matrix(pbmc@assays$RNA@counts)[1:4,1:4]Pvals <- scDEA_individual_methods(raw.count =as.matrix(pbmc@assays$RNA@counts), #此处必须把稀疏矩阵转为常见的矩阵才可以运行----cell.label = pbmc@meta.data$group )combination.Pvals <- lancaster.combination(Pvals, weight = TRUE, trimmed = 0.2)adjusted.Pvals <- scDEA.p.adjust(combination.Pvals, adjusted.method = "bonferroni")
在我自己的数据种,这段代码也是可以运行的哦
后记
1 作者说scDEA这种方法可以减少一类错误,减少细胞数目的影响,减少批次效应的影响......但是,我跑完了他的整个流程,感觉就是真的这么厉害吗

2 另外,为啥实例教程使用hvg呢,而不是使用整个counts?
3 最后,运行的时间有点长啊....

![]()
看完记得顺手点个“在看”哦!
这篇关于scDEA一键汇总12种单细胞差异分析方法 DESeq2、edgeR、MAST、monocle、scDD、Wilcoxon的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!






