本文主要是介绍生物信息数据格式:bed格式,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
文章目录
- BED format(基因组的注释文件)
- 基本列
- 附加列
- 示例
- [Bedtools简介](https://bedtools.readthedocs.io/en/latest/index.html)
- 下载安装
- 演示版的bed文件 (demo.bed)
- 我们的基因组文件(genome.txt)
- [bedtools slop](http://bedtools.readthedocs.io/en/latest/content/tools/slop.html)
- 与GTF的关系
BED format(基因组的注释文件)
用来描述注释的数据。BED线有3个要求的字段(基本列)和9个额外的字段(附加列)
基本列
必不可少的
-
chrom 即chrom 或者scaffold 名称
-
chromStart Feature在chrom中的起始位置(前坐标),chrom的第一个碱基的坐标是0,chromStart如果等于2,其实表示的是第三个碱基,feature包含这个碱基
-
chromEnd feature在chrom中的终止位置(后坐标),chromEnd如果等于5,其实表示的是第六个碱基之前的碱基,feature不包含5这个碱基
详细见https://bedtools.readthedocs.io/en/latest/content/general-usage.html
如下FASTA格式的序列
>chr1
ATGCTTT
对应的bed文件就是:
BED file
chr1 2 5
如果用fastaFromBed提取,那么你能得到的序列是GCT(2号到5号之前的base,第一个base是0号)
附加列
-
name #feature 的名字
-
score 0到1000的分值,如果track线在注释时属性设置为1,那么这个分值会决定现示灰度水平,数字 越大,灰度越高。下面的这个表格显示Genome Browser
-
strand 定义链的’’+” 或者”-”
-
thickStart #feature的起始
-
thickEnd #feature的终止
-
itermRgb R, G, B (eg. 255, 0, 0), 如果track line itemRgb属性是设置为’On”, 这个RBG 值将 决 定数据的显示的颜色在BED 线。
-
blockCount #exon个数
-
blockSize #每个exon的大小
-
blockStarts #以chromStart为起点的各个exon的起始点
示例
BED3
A BED file where each feature is described by chrom, start, and end
chrom start end
chr1 11873 14409
BED4
A BED file where each feature is described by chrom, start, end, and name
chrom start end name
chr1 11873 14409 uc001aaa.3
BED5
A BED file where each feature is described by chrom, start, end, name, and score
chrom start end name score
chr1 11873 14409 uc001aaa.3 0
BED6
A BED file where each feature is described by chrom, start, end, name, score, and strand
chrom start end name score strand
chr1 11873 14409 uc001aaa.3 0 +
BED12
A BED file where each feature is described by all twelve columns listed above
.................
Bedtools简介
下载安装
cd ~/local/app/
curl -OL https://github.com/arq5x/bedtools2/releases/download/v2.22.0/bedtools-2.22.0.tar.gz
tar zxvf bedtools-2.22.0.tar.gz
cd bedtools2
make
ln -sf ~/local/app/bedtools2/bin/bedtools ~/bin/bedtools
演示版的bed文件 (demo.bed)
vim demo.bedKM034562 100 200 one 0 +
KM034562 400 500 two 0 -
我们的基因组文件(genome.txt)
vim genome.txt
KM034562 18959
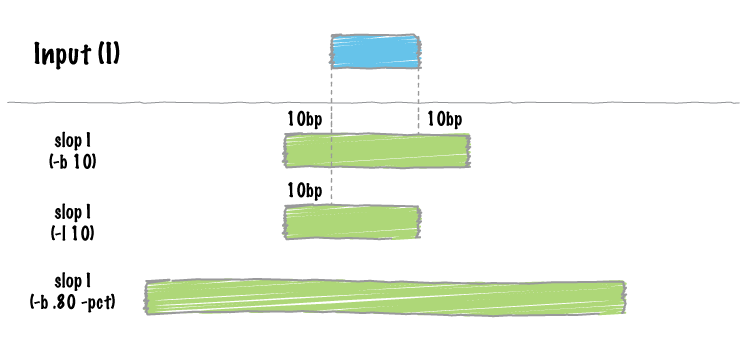
bedtools slop
restrict the resizing to the size of the chromosome
- 参数 -b 增加两端的长度
- 参数 -pct :片段的长度100bp ,-b 0.1 ,会使两端的长度增加10bp
bedtools slop -i demo.bed -g genome.txt -b 10
bedtools slop -i demo.bed -g genome.txt -b 0.1 -pct
KM034562 90 210 one 0 +
KM034562 390 510 two 0 -
- 参数 -l 增加开始端的长度
bedtools slop -i demo.bed -g genome.txt -l 10 -r 0
KM034562 90 203 one 0 +
KM034562 390 503 two 0 -
- 参数 -r 增加末端的长度
bedtools slop -i demo.bed -g genome.txt -l 10 -r 3
KM034562 90 203 one 0 +
KM034562 390 503 two 0 -
- 有链特异性的运算
- 参数 -s 对正链无影响,对于负链 -l 10 不再是增加开始端的长度,而是增加末尾端的长度,而 -r 3 不再是增加末端的长度,而是增加开始端的长度
bedtools slop -i demo.bed -g genome.txt -l 10 -r 3 -s
KM034562 90 203 one 0 +
KM034562 397 510 two 0 -
- 参数 -b
bedtools slop -i demo.bed -g genome.txt -b 20000
KM034562 0 18959 one 0 +
KM034562 0 18959 two 0 -
示意图 :
与GTF的关系
genomic features通常使用Browser Extensible Data (BED) 或者 General Feature Format (GFF)文件表示,用UCSC Genome Browser进行可视化比较。 Bed文件和GFF文件最基本的信息就是染色体或Contig的ID或编号,然后就是DNA的正负链信息,接着就是在染色体上的起始和终止位置数值。
两种文件的区别在于,BED文件中起始坐标为0,结束坐标至少是1,; GFF中起始坐标是1而结束坐标至少是1。
把BED转成对应的GFF
这并非是真的正确地把BED转成GFF
cat demo.bed | bioawk -c bed '{print $chrom, ".", ".", $start+1, $end, $score, $strand, ".", "." }' > demo.gff
less demo.gff
KM034562 . . 101 200 0 + . .
KM034562 . . 401 500 0 - . .
它与其他格式可以很好地协同工作!
bedtools slop -i demo.gff -g genome.txt -l 10 -r 0 -s
KM034562 . . 91 200 0 + . .
KM034562 . . 401 510 0 - . .
更多用法详见
这篇关于生物信息数据格式:bed格式的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!






