本文主要是介绍NAR | 加拿大麦吉尔大学夏建国组更新微生物组数据分析网站 MicrobiomeAnalyst 2,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
MicrobiomeAnalyst 2.0:全面的微生物组数据统计、功能和整合分析
MicrobiomeAnalyst 2.0: comprehensive statistical, functional and integrative analysis of microbiome data

Article,2023-5-3,Nucleic Acids Research,[IF 14.9]
DOI:https://doi.org/10.1093/nar/gkad407
原文链接:https://academic.oup.com/nar/advancearticle/doi/10.1093/nar/gkad407/7160190?login=false
第一作者:Yao Lu
通讯作者:Jianguo Xia (夏建国)
合作作者:Guangyan Zhou;Jessica Ewald;Zhiqiang Pang;Tanisha Shiri
主要单位:加拿大麦吉尔大学微生物与免疫系 (Department of Microbiology and Immunology, McGill University, Quebec, Canada)
加拿大麦吉尔大学寄生虫学研究所 (Institute of Parasitology, McGill University, Quebec, Canada)
加拿大麦吉尔大学动物科学系(Department of Animal Science, McGill University, Quebec, Canada)
- 摘要 -
微生物组研究在生物医学、农业和环境科学等学科中已经非常常见,其目标包括多样性分析、功能特性描述和转录应用等多个方面。由此产生的复杂的,常常涉及多组学数据的数据集则需要强大而又用户友好的生物信息学工具来揭示关键模式、重要生物标志物和潜在通路。在这里,我们介绍MicrobiomeAnalyst 2.0,它可以支持微生物组研究中常见的数据输出的全面统计分析、可视化、功能注释和整合分析。与上一版本相比,MicrobiomeAnalyst 2.0增加了三个新模块:(i) 原始数据处理模块,用于处理扩增子数据和进行分类注释,并直接与丰度数据分析模块(Marker Data Profiling module)对接,用于下游统计分析;(ii) 微生物代谢组学分析模块,通过联合分析微生物组和代谢组学数据,帮助解析群落组成与代谢通路之间的关联;(iii) 统计整合分析模块,通过整合多个研究的数据集,帮助识别一致的特征。其他重要改进包括增加对多因素差异分析的支持,提供交互式可视化界面以展示常见的图形输出,更新功能预测和相关性分析方法,并基于最新文献扩展了分类库。这些新功能通过最近的一项Ⅰ型糖尿病研究的多组学数据进行了演示。MicrobiomeAnalyst 2.0可以免费访问,网址是https://microbiomeanalyst.ca 。
- 引言 -
在过去的十年中,微生物组研究在各个学科领域中增长迅速,明显趋势是利用多组学技术对群落进行全面的表征。微生物组现在被认为是人类健康和可持续农业中的关键因素。强大的生物信息学流程和工具不断被开发和更新,以帮助分析日益复杂的数据集。MicrobiomeAnalyst 1.0版旨在为实验室研究人员提供一个用户友好的基于Web的平台,用于对常见的丰度分布和分类学特征进行全面的探索性分析(11)。自2017年发布以来,MicrobiomeAnalyst根据用户反馈不断更新,并于2020年发布了详细的分析教程。根据Google Analytics的数据,Microbiome Analyst公共服务器在过去12个月内处理了来自全球超过30,000名用户提交的超过125,000个作业。
微生物组数据分析在概念上与其他组学数据分析工作流程类似,包括原始数据处理、统计分析和功能解释这三个典型阶段。然而,在实践中,微生物组数据显示出更高的异质性,特别是在个体间和群体间存在很大差异,导致了包括零膨胀(Zero Inflation)、组成性 (Compositionality)和过度离散性 (Overdispersion)在内的统计问题。这些特点促使了各种分析方法的发展,导致了对于不精通统计学或编程的研究人员来说,分析方法的选择变得具有挑战性。标记基因数据分析已经从传统的操作性分类单元 (OTUs)转变为基于其独特生物序列进行鉴定的高分辨率引物序列变体 (ASVs)。使用ASVs不仅减少了与序列聚类相关的计算瓶颈,还促进了跨不同研究的比较分析。在差异丰度分析中,几个基准研究显示了专门针对微生物组数据开发的方法之间存在的不一致性,并且这种常见的RNA测序分析方法表现出稳健性和良好的性能。最后,对于易于使用但同时具有灵活性的工具的需求不断增长,这些工具可以考虑复杂的元数据,并支持微生物组学研究的多组学整合(19-21)。
为了跟上最近微生物组学研究中的进展和不断演变的数据分析需求,我们对MicrobiomeAnalyst平台进行了重大更新,包括三个新模块:(i)一个用于标记基因数据的原始数据处理模块,直接与下游统计分析对接;(ii)一个微生物组代谢组学模块,用于微生物组和代谢组学数据联合分析,以及(iii) 一个用于多标记基因数据集的统计整合分析模块。我们还对先前的模块进行了重大更新,包括对复杂元数据的支持(元数据编辑器、连续元数据和多因子比较分析),增强的统计方法(功能预测和相关网络分析),新的交互式可视化(堆叠条形图、热图和KEGG代谢网络),以及基于最新文献扩展的分类群库。MicrobiomeAnalyst 2.0可以免费在microbiomeanalyst.ca上获得。它包含全面的教程,并配备了一个专门的用户论坛(omicsforum.ca)。底层的MicrobiomeAnalystR包也已发布(https://github.com/xia-lab/MicrobiomeAnalystR),以促进透明和可重复的分析。
- 结果 -
软件描述和方法
Program description and methods
MicrobiomeAnalyst 2.0的工作流程包括四个主要步骤(图1)。它支持常见的输入类型,包括16S、18S rRNA基因或内转录间隔(ITS)区域的原始引物测序数据,由标记基因或宏基因组生成的计数表,配对的微生物组和代谢组学数据表或列表,来自兼容研究的多个标记基因计数表,或者分类学特征。上传后,所有输入数据都遵循相同的通用数据处理、方法选择和结果探索的工作流程。在每个步骤中,都提供了全面的选项和分析支持。在接下来的章节中,我们将主要关注版本2.0中引入的新功能或改进的功能。
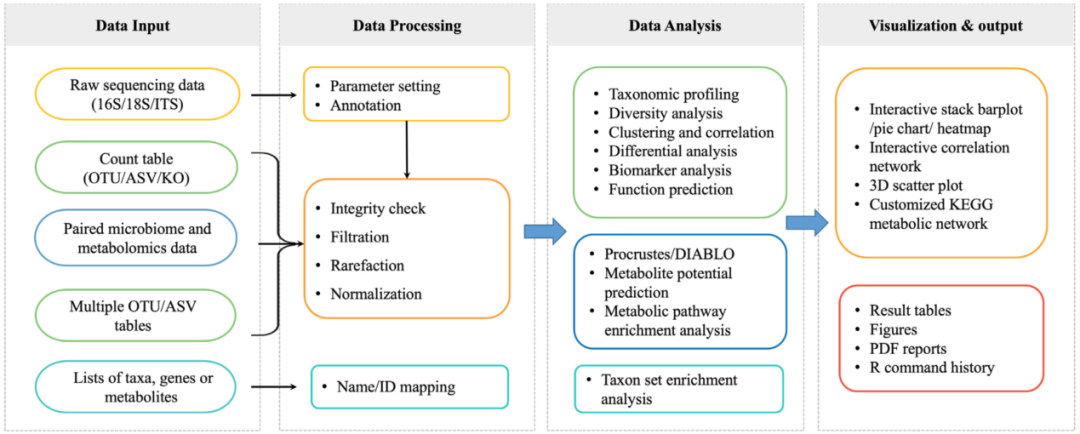
图一:MicrobiomeAnalyst 2.0 工作流程
扩增子测序数据处理
Amplicon sequencing data processing
扩增子测序为揭示人类疾病的发展和进展提供了许多见解。它已成为研究微生物组复杂性和多样性的一种无处不在的方法。与宏基因组测序相比,扩增子测序既具有成本效益,又在计算上更高效,尤其适用于具有许多低丰度物种的高度异质性群落。在下游分析之前,原始读数需要首先被处理成OTUs或ASVs。现存许多用于原始数据处理的工具,包括QIIME2、Mothur和DADA2。然而,使用这些工具需要命令行技能。MicrobiomeAnalyst 2.0引入了一个新模块,基于成熟的DADA2工作流程,提供了一个自动化的流程,用于处理扩增子数据。用户可以上传来自16S/18S/ITS测序的单端或双端压缩FASTQ文件(.gz或.zip)。进一步的下游统计分析还需要一个以纯文本格式(.txt或.csv)的元数据文件。该工作流程包括过滤、去重、样本推断、嵌合体识别和双端读长合并。MicrobiomeAnalyst 2.0提供了一个参数选择页面,允许用户根据质量控制图形输出来调整处理参数。分类注释基于几个参考数据库,包括16S测序的SILVA(v138)、Greengenes(13.8)和RDP(release 11.5)数据库,ITS测序的UNITE数据库,以及18S测序的SILVA(v132)。当原始数据定量处理完成后,将为每个样本生成摘要图形和详细处理信息。生成的ASV和分类表可以下载或直接通过点击模块重定向按钮用作标记数据分析的输入。
来自多个微生物代谢组研究数据的整合分析
Integrative analysis for data from microbiome metabolomics studies
代谢物在微生物之间和其与宿主之间的交流和相互作用中起着关键作用。代谢组学在最近的微生物组研究中越来越多地被用于连接微生物群落组成和改变的代谢过程的表型特征。然而,整合高维微生物组和代谢组学数据仍然是一个主要挑战。为了解决这个问题,MicrobiomeAnalyst 2.0引入了一个新模块,允许用户探索微生物组装配与其代谢产物之间的关系。
用户可以上传配对的丰度表或配对的列表。对于微生物组数据,输入特征可以是OTUs、ASVs或KEGG Orthologs (KOs)。对于代谢组学数据,输入特征可以是代谢物(靶标代谢组学)或LC-MS峰(非靶标代谢组学)。对于表格输入,根据输入数据类型提供了不同的数据过滤和归一化方法。统计比较微生物组和代谢组学数据分别使用了MaAsLin2和limma方法。这两种方法都依赖于通用线性模型来确定组学特征与复杂元数据之间的关联,并支持协变量调整。列表输入直接提交给名称匹配步骤,以准备进行进一步的整合分析。微生物组-代谢整合分析通过降维、代谢网络分析和相关性分析三种方法实现。
降维 (Dimensionality reduction):为了揭示微生物组和代谢组学数据集之间的整体模式,采用了两种强大的降维方法,Procrustes分析(PA)和用于潜在成分的生物标记发现的数据整合分析(DIABLO)。PA是一种无监督方法,通过旋转一个数据集的轴,直到达到最大相似度,将两个数据集的主成分叠加在一起。DIABLO是一种有监督方法,旨在识别能够最大程度解释单个数据和它们与感兴趣元数据的协方差的多组学成分。相关结果以交互式三维散点图的形式呈现。用户可以在得分图、载荷系数图和双标图之间切换,以可视化高级趋势,突出显示具有不同元数据的结果,或者识别感兴趣的特征。
代谢网络分析(Metabolic network analysis):该模块旨在提供基于上传的微生物组装配数据中存在的分类或KOs的代谢分析。用户可以根据在微生物组数据中检测到的具有统计学意义的分类或所有分类,自定义全局代谢网络。或者,用户可以选择基于聚合的微生物代谢网络或其与宿主代谢网络的组合的通用(未过滤)代谢背景。使用两种成熟的算法——mummichog和globaltest分别对LC-MS峰和其他特征进行富集分析。结果在交互式全局代谢网络中可视化,其中节点表示代谢物,边表示酶反应,不符合特定研究的微生物潜力或KO轮廓的反应会变灰。用户可以在表格中点击任何富集的代谢通路名称,以在网络上突出显示相应的代谢物或KO。用户还可以直接点击网络中的节点(代谢物),以圆形图的形式显示与之最相关的微生物。
微生物组-代谢组关联分析 (Microbiome-metabolome correlation analysis): 该模块支持统计、基于模型的和整合的相关性分析。对于统计相关性分析,默认选项是基于距离的相关性方法,可以检测线性和非线性相关性。其他选项包括Pearson、Kendall和Spearman相关性及其相应的偏相关性。结果以交互式热图的形式进行总结。两两相关性分析通常会导致大量的假阳性,使得生物学解释变得困难。为了解决这个问题,我们实施了一种基于超过5000个高质量基因组规模代谢模型(GEMs)的基于模型的相关性,以提供微生物分类与其代谢物之间的概率热图。最后,用户可以选择叠加统计相关性和基于模型的相关性热图,以整合数据驱动和知识驱动的证据流。
不同微生物研究间的统计整合分析(Statistical meta-analysis across multiple microbiome studies): 由于实验设计、分析方法和定量评估的差异性,在不同的微生物组研究中实现可重复的特征非常具有挑战性。统计整合分析模块旨在提供一个框架,用于整合具有相同表型的多个标记基因研究的数据,以帮助识别稳健且可重复的特征。数据上传和处理步骤与单个标记基因数据分析工作流程类似,额外添加了一个验证步骤,以确保所有数据集和元数据的一致性。在处理完成后,执行批次校正以调整潜在的技术变异,提高不同微生物组研究之间的可比性。在此步骤之后,提供三种整合分析策略:可视化探索、生物标志物整合分析和多样性整合分析。
可视化探索 (Visual exploration): 该方法提供堆叠面积/柱状图和主坐标分析(PCoA)图,以概述高级模式,同时允许用户调查样本级细节。堆叠面积/柱状图提供了在所有数据集中对分类群丰度进行样本级概况,以更好地理解分类组成,而PCoA则提供了样本和数据集之间微生物组成的相似性/差异性的概述。请注意,之前的“投射到公共数据”模块已迁移到此页面上。
生物标记物整合分析 (Biomarker meta-analysis): 该方法的目标是将个别数据集的差异丰度测试结果整合起来,以识别与感兴趣表型相关的常见微生物标志物。该方法由两部分组成:首先在各个数据集中使用多元线性回归进行丰度测试,然后使用基于MMUPHin R软件包的随机效应模型集成效应大小。结果以条形图的形式呈现,显示前几个显著特征,并包含详细的表格,其中包含各个研究中所有特征的统计摘要信息。
丰度分析 (Diversity meta-analysis): 该方法整合了数据集中的α和β多样性指数。为每个研究计算常见的α多样性指数,并使用箱线图和森林图查看实验组之间指数的比率。通过对每个研究的常见距离矩阵进行主坐标分析,整合了β多样性指数。可以使用多个统计检验(如PERMANOVA、ANOSIM、PERMDISP和MiRKAT)来衡量表型对群落组成的影响的显著性。α和β多样性整合分析提供图形摘要和详细表格。
其他特性
Other features
复杂元数据的多因素分析(Multi-factor analysis for complex metadata):随着微生物组数据集的规模越来越大,实验设计也变得越来越复杂,因此元数据也变得更加复杂。此外,在观察性研究中,复杂元数据尤为重要,通常同时测量连续和分类协变量。因此,我们在MicrobiomeAnalyst 2.0中投入了大量的努力来增强对元数据的支持。在数据完整性检查页面上实现了一个元数据面板,用户可以检查和编辑元数据变量,包括指定它们是连续的还是分类的。用户还可以指定分类元数据的组标签顺序。基于通用线性模型,使用MaAsLin2 R软件包实现了多因素比较工具。用户指定感兴趣的主要元数据,并可以包括年龄、性别或技术因素等协变量进行调整。协变量可以被建模为固定效应或随机效应。针对每个特征,使用包含主要元数据和所有协变量的线性模型进行拟合,然后从模型中提取用于主要元数据的统计信息。
改进的相关分析和功能预测(Improved correlation analysis and function prediction):基于领域的最新发展,更新了几个用于标记基因分析的功能。MicrobiomeAnalyst 2.0现在提供了七种相关方法供用户探索微生物之间的关系,包括最近的Sparse Estimation of Correlations among Microbiomes (SECOM)方法,该方法提供了微生物之间的线性和非线性关系度量。对于从16S rRNA基因丰度表预测功能能力,以前的版本基于GreenGenes和SILVA分类注释分别提供了PICRUSt和Tax4Fun。在2.0版本中,我们更新了PICRUSt的数据库,支持对>200,000个OTU进行∼7000个KO的注释。还可以使用Tax4Fun2,允许用户直接从ASV序列预测潜在功能。
扩展的大数据探索可视化(Enhanced visualizations for large data exploration):我们为堆叠条形/面积图和聚类热图实现了交互式绘图,这些功能是我们的用户最常请求的用于大型数据集的可视化探索功能。支持鼠标悬停和缩放效果,允许用户获取感兴趣特征/模式的详细信息。另一个改进是更新了KEGG代谢网络(版本105.0),以改进可视化和功能分析。
扩展的分类数据库(Expanded taxon set libraries):创建了Taxon Set Enrichment Analysis (TSEA)模块,允许研究人员识别以其共享功能或与特定表型相关联的分类标志物,以促进数据解释和假设生成。TSEA针对感兴趣的分类集合库执行超几何测试,以从输入的微生物特征列表中检测最常表示的标志物。在2.0版本中,我们整合了来自流行数据库(如gutMDisorder,GIMICA和MiMeDB)的数据,并扩展了表型特征的列表,包括与免疫反应相关的102个微生物组特征,77个微生物组-代谢物关联,55个与癌症相关的分类集合以及与药物治疗相关的137个分类集合。为了提高统计能力和生物学相关性,我们进一步整合了至少有四个或更多微生物成员的分类集合。分类集合库现在总共包含611个与宿主内在特征相关的特征,696个与饮食、药物和生活方式相关的宿主外在特征,500个与环境特征相关的特征,以及>700个与单核苷酸多态性(SNP)相关的分类集合。
实例研究
Case study
为展示MicrobiomeAnalyst 2.0的新功能,我们利用了一项最近的1型糖尿病(T1D)研究。T1D是一种自身免疫性疾病,会导致胰岛β细胞破坏和胰岛素缺乏。先前的研究表明,T1D可以由多种因素引起,如遗传易感性、病毒感染、饮食成分以及肠道微生物组。该研究的目标是调查在患有和未患T1D的人群中微生物群落的变化对其影响。进行了16S标记基因测序和基于LC-MS的代谢组学分析。使用原始论文提供的检索号,我们从NCBI Sequence Read Archive(SRA)数据库下载了原始测序数据,并从MetaboLights获取了代谢物浓度表。使用我们的DADA2流程对原始数据进行处理,得到ASV丰度表和分类注释。结果基于Tax4Fun2的预测进行了功能分析。然后,通过将代谢物丰度与ASV计数数据或KO丰度表集成进行了两种类型的共同分析。以属水平为例,解释了图2中呈现的结果。
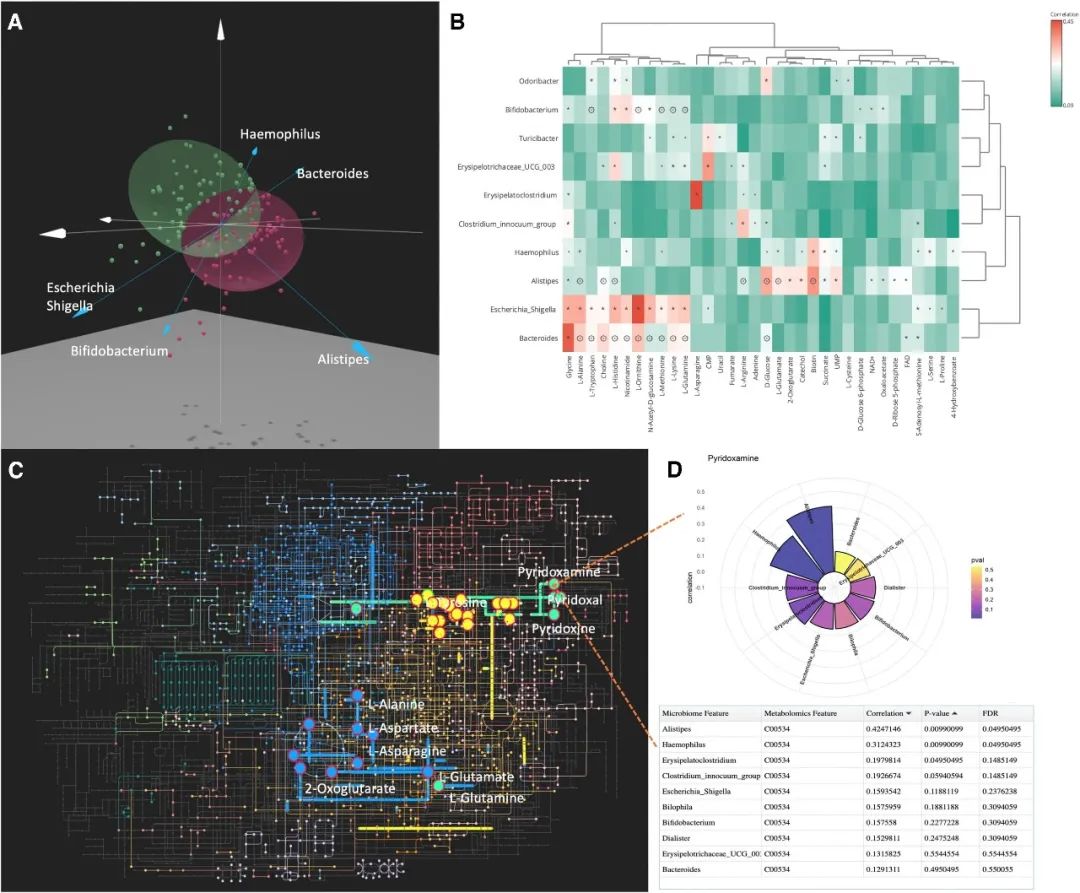
图2. 示例
(A) 在3D散点图中呈现的DIABLO结果。
(B) 统计相关结果与基于模型的相关热图叠加。本分析中使用了通过调整后P值0.1的阈值的特征。颜色渐变表示统计相关性,星号显示通过原始P值0.05进行了统计显著性过滤的相关性。菱形表示这些相关性也被基于GEM的预测模型所预测。
(C) 基于KOs和代谢物的通路富集结果(黄色:酪氨酸代谢;绿色:维生素B6代谢;蓝色:丙氨酸、天冬氨酸和谷氨酸代谢)。
(D) 圆形图与下方显示选定代谢物(pyridoxamine)最相关的分类群的详细表格。该结果是通过点击相应节点获得的。
图2A展示了DIABLO双标图结果以3D散点图形式呈现。T1D和对照组的组成在一定程度上重叠,与原始研究结果一致。观察到一些微生物分类群,如Bacteroides和Alistipes,与前几个主要成分相关。我们假设这些微生物通过特定代谢物在T1D和非糖尿病受试者之间产生分离效应。通过叠加的热图展示了详细的微生物-代谢物相关性(图2B)。在比较分析中,仅使用调整后P值<0.1的特征。统计相关性分析使用了基于距离的方法,选择了AGORA数据库进行基于GEM的预测结果。在显著性水平0.05的筛选下,我们可以观察到两种方法均显示Bacteroides与葡萄糖、谷氨酰胺和几种氨基酸显著相关。Alistipes也与一组不同的氨基酸相关,与DIABLO分析结果一致。尽管Bacteroides在原始论文中未被鉴定为生物标志物,但其他研究表明它与饮食有关,并且是早期自身抗体产生的危险因素。大多数研究集中在T1D中Bacteroides物种的组成变化,而未与功能相关联。我们的分析显示了与Bacteroides显著相关的代谢物,提示它可能通过“丙氨酸、天冬氨酸和谷氨酸代谢”影响T1D。图2C展示了利用全局检验方法对代谢物和KOs在KEGG代谢途径上的富集分析的综合结果。两个数据集均检测到了几个途径(在图2C左侧突出显示),包括“维生素B6代谢”、“酪氨酸代谢”和“丙氨酸、天冬氨酸和谷氨酸代谢”。T1D和对照组之间差异的途径可以在网络中以不同颜色显示。点击网络中相应节点可以可视化与每个代谢物相关的分类群。例如,缺乏pyridoxamine可能会影响胰岛素信号传导。图2D展示了与pyridoxamine最相关的前10个属,例如Alistipes。我们注意到,在T1D组和对照组之间,“维生素B6代谢”内的代谢物没有显著差异,然而富集分析仍然可以在途径水平上识别出变化。
实例研究
Implementation
MicrobiomeAnalyst 2.0的Web界面是基于JavaServer Faces框架和PrimeFaces库(https://www.primefaces.org,版本12.0.0)实现的。统计功能和图形功能是使用R语言(版本4.2.2)实现的,并可以从GitHub存储库(https://github.com/xia-lab/MicrobiomeAnalystR)免费获取。为了适应不断增长的用户流量和计算需求,该系统部署在Google Cloud实例上,并通过在McGill数据中心托管的第二个计算节点进行负载平衡。对于原始数据处理,作业提交和调度基于Simple Linux Utility for Resource Management(SLURM)系统进行。
与其他工具比较
Comparison with other tools
已经有很多基于Web的微生物组数据分析工具。在这里,我们将MicrobiomeAnalyst 2.0与其他四个工具以及先前版本进行了比较。表1总结了每个工具的主要特点。专用于处理和存档原始序列数据的流行工具,例如基因组学快速注释子系统技术(MG-RAST)和MGnify(以前称为EBI Metagenomics)在此处未列出。MicrobiomeAnalyst 1.0是为了满足统计分析需求而开发的,提供了全面的功能列表和出版就绪的图形。类似的工具包括微生物种群结构分析(VAMPS)、Namco和MIAN。然而,只有新建的Namco具有与MicrobiomeAnalyst 2.0相当数量的分析选项。全球宏基因组目录(gcMeta)旨在注释和分析标记基因和随机宏基因组的原始数据,并由大量多组学研究支持,然而它提供非常有限的分析方法,并且没有相应的整合分析方法。最后,微生物组和代谢组学数据的综合分析已经满足了微生物社区的紧迫需求。总体而言,MicrobiomeAnalyst 2.0是最全面的基于Web的平台,可进行用户友好且流畅的微生物组数据分析和解释。
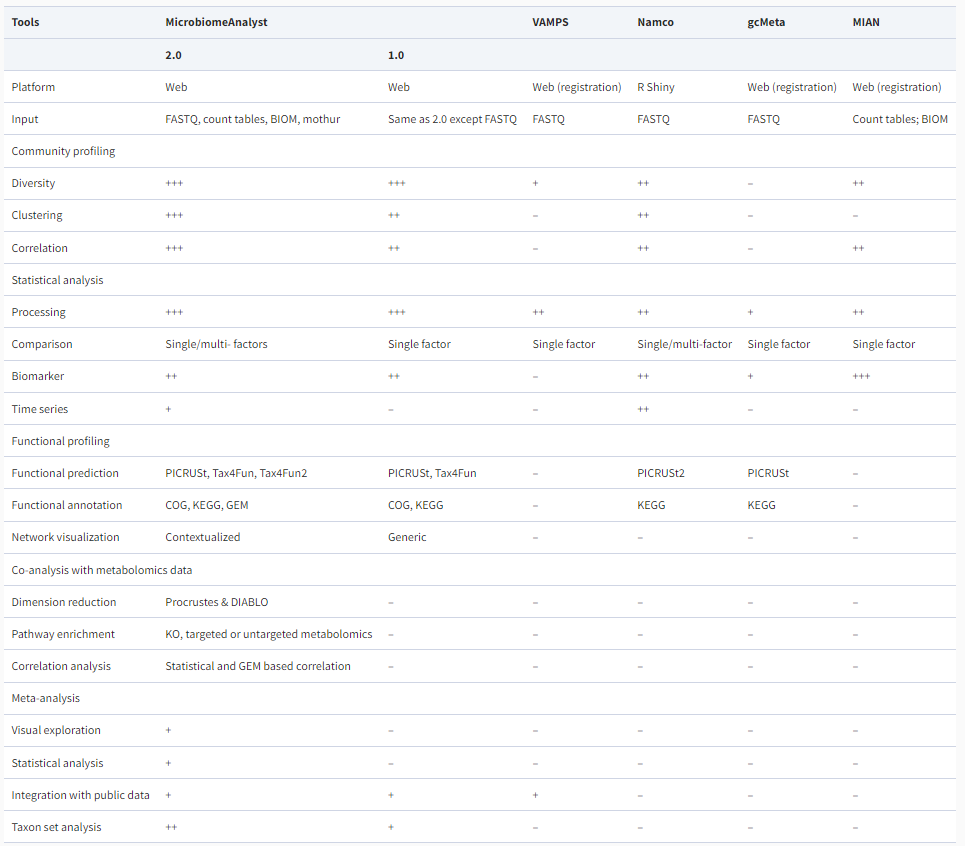
表格1.MicrobiomeAnalyst 2.0与其他网络工具的比较。
用于特征评估的符号为‘-’表示缺失,‘+’表示更定量的评估(更多的‘+’表示更好的支持,例如更好的可视化和提供更多选项)。
- 结论 -
MicrobiomeAnalyst 2.0是为满足微生物组数据分析的快速发展需求而开发的。它提供了一个基于Web的平台,使研究人员能够轻松地探索和理解他们的数据。为了跟上最新的发展,我们更新了功能注释、分类集富集分析的库,并嵌入了几种最新的统计方法,以增强1.0版本中开发的模块。在2.0版本引入的三个新模块中,MicrobiomeAnalyst现在支持从原始数据处理到下游统计和功能分析的标记基因数据的流畅分析。它还支持对成对的微生物组-代谢组学数据集以及多个标记基因计数表的整合分析。我们的案例研究表明,MicrobiomeAnalyst 2.0可以从复杂的数据集中提取信息,揭示与T1D相关的微生物和代谢物之间的潜在机制联系。由于互联网带宽和大量用户流量的限制,公共服务器目前在每个分析会话中将计数表的最大文件大小限制为50MB,原始序列文件限制为100个。我们建议计划进行大规模数据分析的研究人员使用MicrobiomeAnalystR软件包。在未来,我们的目标是支持更多类型的分析,如单细胞数据分析或在宿主遗传背景下的因果推断。
参考文献
Yao Lu, Guangyan Zhou, Jessica Ewald, Zhiqiang Pang, Tanisha Shiri, Jianguo Xia. MicrobiomeAnalyst 2.0: comprehensive statistical, functional and integrative analysis of microbiome data[J]. Nucleic Acids Research, 2023: gkad407.
- 作者简介 -
第一作者

巴黎第四大学
Yao Lu
硕士
Yao Lu在2011年获得中国药科大学药物工程学士学位。她于2014年获得北京协和医学院微生物与生化药学硕士学位,然后在2019年获得巴黎第四大学生物医学工程硕士学位。她的研究兴趣是功能注释和微生物组与代谢组学的整合。
通讯作者

麦吉尔大学
夏建国
助理教授
夏建国, 2015年加入麦吉尔大学担任助理教授。他目前的研究重点是整合大数据分析和高通量组学技术以理解基因-环境相互作用,并应用于代谢组学、微生物组学、毒物基因组学和传染病研究。
猜你喜欢
iMeta简介 高引文章 高颜值绘图imageGP 网络分析iNAP
iMeta网页工具 代谢组MetOrigin 美吉云乳酸化预测DeepKla
iMeta综述 肠菌菌群 植物菌群 口腔菌群 蛋白质结构预测
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature
系列教程:微生物组入门 Biostar 微生物组 宏基因组
专业技能:学术图表 高分文章 生信宝典 不可或缺的人
一文读懂:宏基因组 寄生虫益处 进化树 必备技能:提问 搜索 Endnote
扩增子分析:图表解读 分析流程 统计绘图
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流快速解决科研困难,我们建立了“宏基因组”讨论群,己有国内外6000+ 科研人员加入。请添加主编微信meta-genomics带你入群,务必备注“姓名-单位-研究方向-职称/年级”。高级职称请注明身份,另有海内外微生物PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。
点击阅读原文,跳转最新文章目录阅读
这篇关于NAR | 加拿大麦吉尔大学夏建国组更新微生物组数据分析网站 MicrobiomeAnalyst 2的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!







