本文主要是介绍WGCNA分析教程五 | [更新版],希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
一边学习,一边总结,一边分享!
往期WGCNA分析教程
-
WGCNA分析 | 全流程分析代码 | 代码一
-
WGCNA分析 | 全流程分析代码 | 代码二
-
WGCNA分析 | 全流程分析代码 | 代码四
关于WGCNA分析教程日常更新
学习无处不在,我们的教程会在原有的基础上不断进行更新和添加新的内容,以及不断完善的分析代码。尽最大的能力,让大家拿到代码后直接上手分析。而不需要调整那些看不懂的参数而烦恼(但这仅限于自己的能力范围之内)。
本次WGCNA教程更新,是基于WGCNA分析 | 全流程分析代码 | 代码一的教程。针对原有教程一,增加模块基因文件输出、各模块Hub基因之间的link文件输出(此文件可以直接输入Cytoscape软件,绘制hub基因直接网络图)、相关性共表网络图和共表达基因与性状的相关性网络图
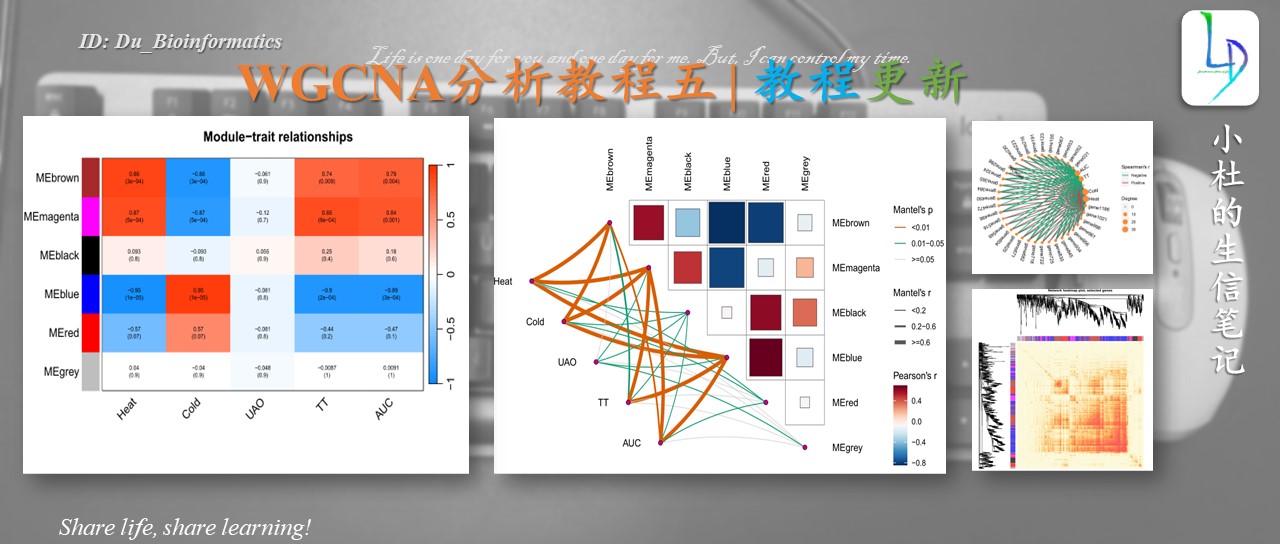
本教程所有图形

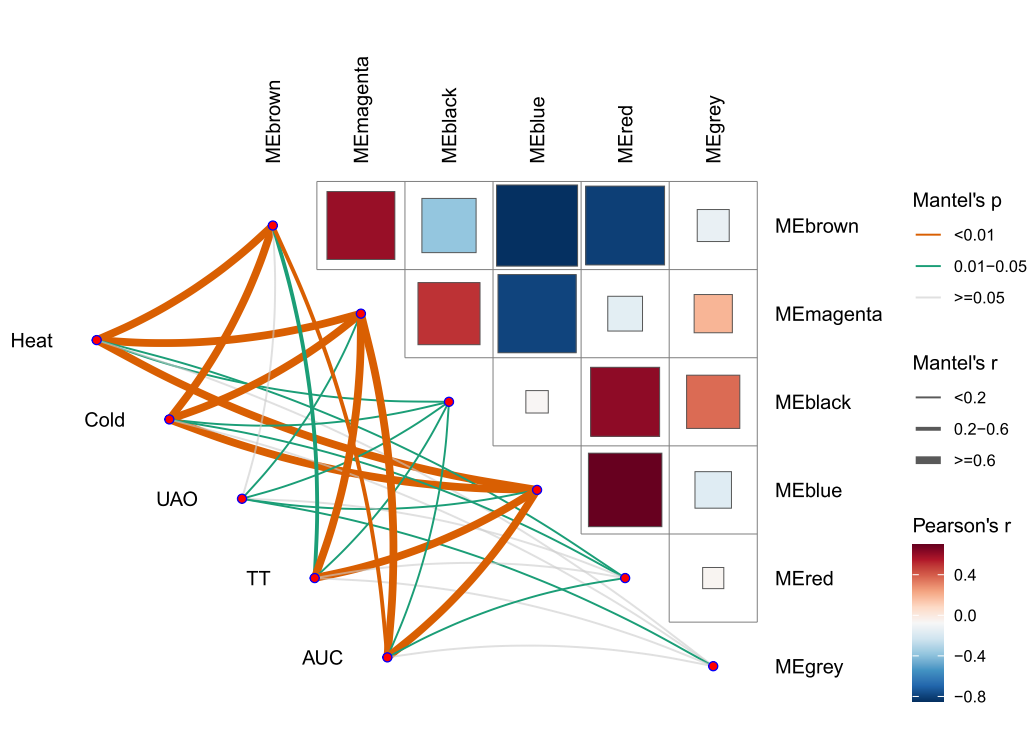

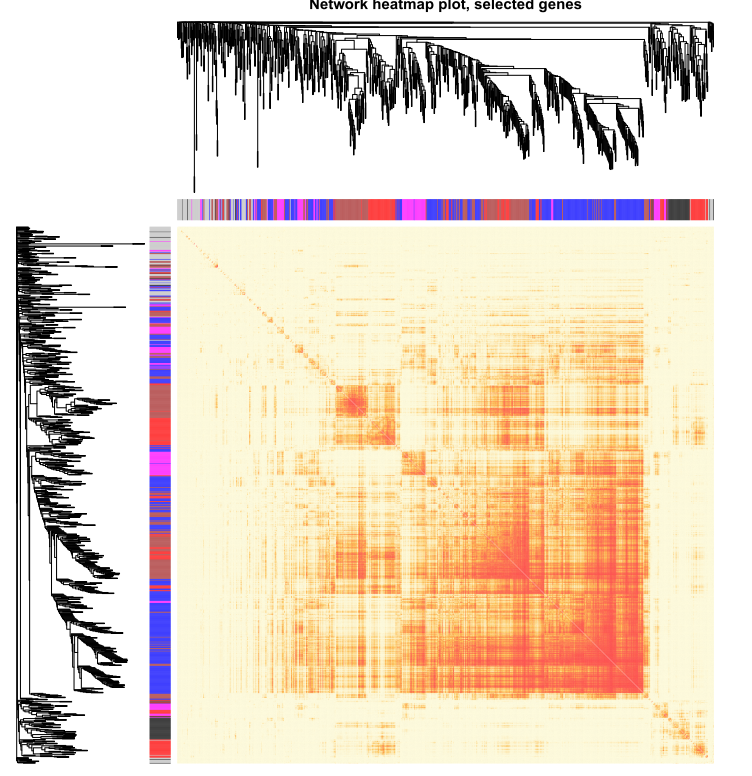

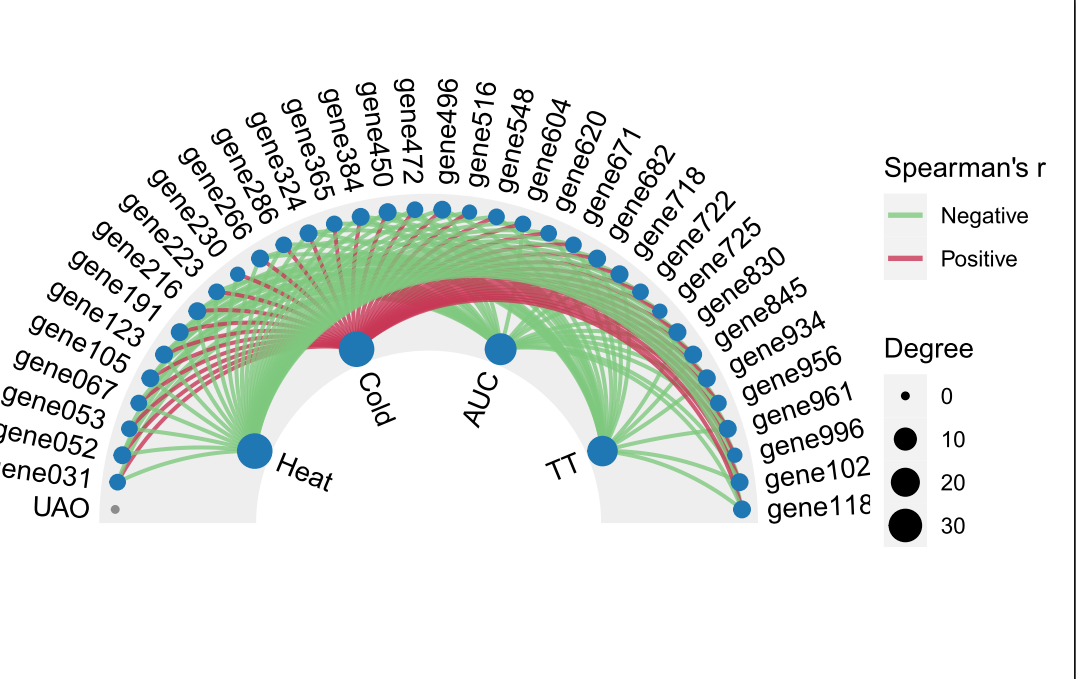
分析代码
关于WGCNA分析,如果你的数据量较大,建议使用服务期直接分析,本地分析可能导致R崩掉。
设置文件位置
setwd("setwd("E:\\小杜的生信筆記\\2023\\20230217_WGCNA\\WGCNA_05")")
加载分析所需的安装包
install.packages("WGCNA")
#BiocManager::install('WGCNA')
library(WGCNA)
options(stringsAsFactors = FALSE)
注意,如果你想打开多线程分析,可以使用一下代码
enableWGCNAThreads()
一、导入基因表达量数据
##'@标准化【optional】
##'@对于数据标准化,根据自己的需求即可,非必要
# WGCNA.fpkm <- log2(WGCNA.fpkm +1 )
# write.csv(WGCNA.fpkm, "ExpData_WGCNA_log2.csv")## 读取txt文件格式数据
WGCNA.fpkm = read.table("ExpData_WGCNA.txt",header=T,comment.char = "",check.names=F)
###############
# 读取csv文件格式
WGCNA.fpkm = read.csv("ExpData_WGCNA.csv", header = T, check.names = F)
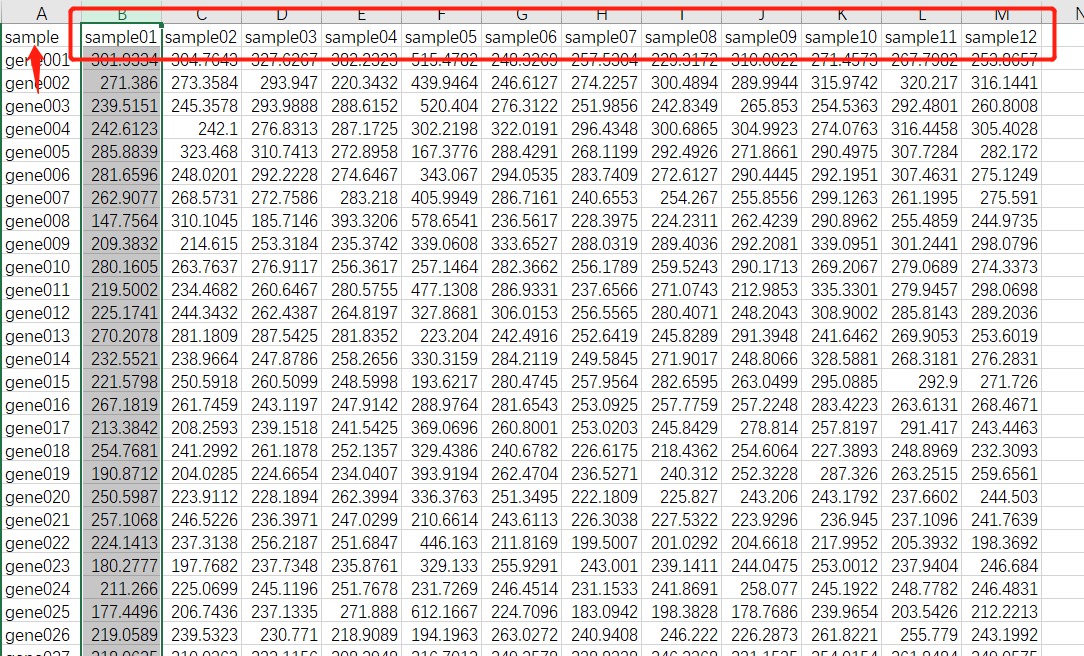
数据处理
dim(WGCNA.fpkm)
names(WGCNA.fpkm)
datExpr0 = as.data.frame(t(WGCNA.fpkm[,-1]))
names(datExpr0) = WGCNA.fpkm$sample;##########如果第一行不是ID命名,就写成fpkm[,1]
rownames(datExpr0) = names(WGCNA.fpkm[,-1])
过滤数据
gsg = goodSamplesGenes(datExpr0, verbose = 3)
gsg$allOK
if (!gsg$allOK)
{if (sum(!gsg$goodGenes)>0)printFlush(paste("Removing genes:", paste(names(datExpr0)[!gsg$goodGenes], collapse = ", ")))if (sum(!gsg$goodSamples)>0)printFlush(paste("Removing samples:", paste(rownames(datExpr0)[!gsg$goodSamples], collapse = ", ")))# Remove the offending genes and samples from the data:datExpr0 = datExpr0[gsg$goodSamples, gsg$goodGenes]
}
过滤低于设定的值的基因
##filter
meanFPKM=0.5 ###--过滤标准,可以修改
n=nrow(datExpr0)
datExpr0[n+1,]=apply(datExpr0[c(1:nrow(datExpr0)),],2,mean)
datExpr0=datExpr0[1:n,datExpr0[n+1,] > meanFPKM]
# for meanFpkm in row n+1 and it must be above what you set--select meanFpkm>opt$meanFpkm(by rp)
filtered_fpkm=t(datExpr0)
filtered_fpkm=data.frame(rownames(filtered_fpkm),filtered_fpkm)
names(filtered_fpkm)[1]="sample"
head(filtered_fpkm)
write.table(filtered_fpkm, file="mRNA.filter.txt",row.names=F, col.names=T,quote=FALSE,sep="\t")
Sample cluster
sampleTree = hclust(dist(datExpr0), method = "average")
pdf(file = "1.sampleClustering.pdf", width = 15, height = 8)
par(cex = 0.6)
par(mar = c(0,6,6,0))
plot(sampleTree, main = "Sample clustering to detect outliers", sub="", xlab="", cex.lab = 2,cex.axis = 1.5, cex.main = 2)
### Plot a line to show the cut
#abline(h = 180, col = "red")##剪切高度不确定,故无红线
dev.off()
不过滤数据
如果你的数据不进行过滤直接进行一下操作,此步与前面的操作相同,任选异种即可。
##'@此步这里我们不过滤,直接使用
## Determine cluster under the line
clust = cutreeStatic(sampleTree, cutHeight = 50000, minSize = 10)
table(clust)
# clust 1 contains the samples we want to keep.
keepSamples = (clust!=0)
datExpr0 = datExpr0[keepSamples, ]
write.table(filtered_fpkm, file="WGCNA.过滤后数据.txt",,row.names=F, col.names=T,quote=FALSE,sep="\t")###
#############Sample cluster###########
sampleTree = hclust(dist(datExpr0), method = "average")
pdf(file = "1.sampleClustering.pdf", width = 6, height = 4)
par(cex = 0.6)
par(mar = c(0,6,6,0))
plot(sampleTree, main = "Sample clustering to detect outliers", sub="", xlab="", cex.lab = 2,cex.axis = 1.5, cex.main = 2)
### Plot a line to show the cut
#abline(h = 180, col = "red")##剪切高度不确定,故无红线
dev.off()
去除离群值
pdf("2_sample clutering_delete_outliers.pdf", width = 6, height = )
par(cex = 0.6)
par(mar = c(0,6,6,0))
cutHeight = 15 ### cut高度
plot(sampleTree, main = "Sample clustering to detect outliers", sub="", xlab="", cex.lab = 2, cex.axis = 1.5, cex.main = 2) +abline(h = cutHeight, col = "red") ###'@"h = 1500"参数为你需要过滤的参数高度
dev.off()
过滤离散样本[optional]
此步可选步骤。
##'@过滤离散样本
##'@"cutHeight"为过滤参数,与上述图保持一致
clust = cutreeStatic(sampleTree, cutHeight = cutHeight, minSize = 10)
keepSamples = (clust==1)
datExpr = datExpr0[keepSamples, ]
nGenes = ncol(datExpr)
nSamples = nrow(datExpr)
dim(datExpr)
head(datExpr)
datExpr[1:11,1:12]
二、导入性状数据
##'@导入csv格式
traitData = read.csv("TraitData.csv",row.names=1)
# ##'@导入txt格式
# traitData = read.table("TraitData.txt",row.names=1,header=T,comment.char = "",check.names=F)
head(traitData)
allTraits = traitData
dim(allTraits)
names(allTraits)
# 形成一个类似于表达数据的数据框架
fpkmSamples = rownames(datExpr)
traitSamples =rownames(allTraits)
traitRows = match(fpkmSamples, traitSamples)
datTraits = allTraits[traitRows,]
rownames(datTraits)

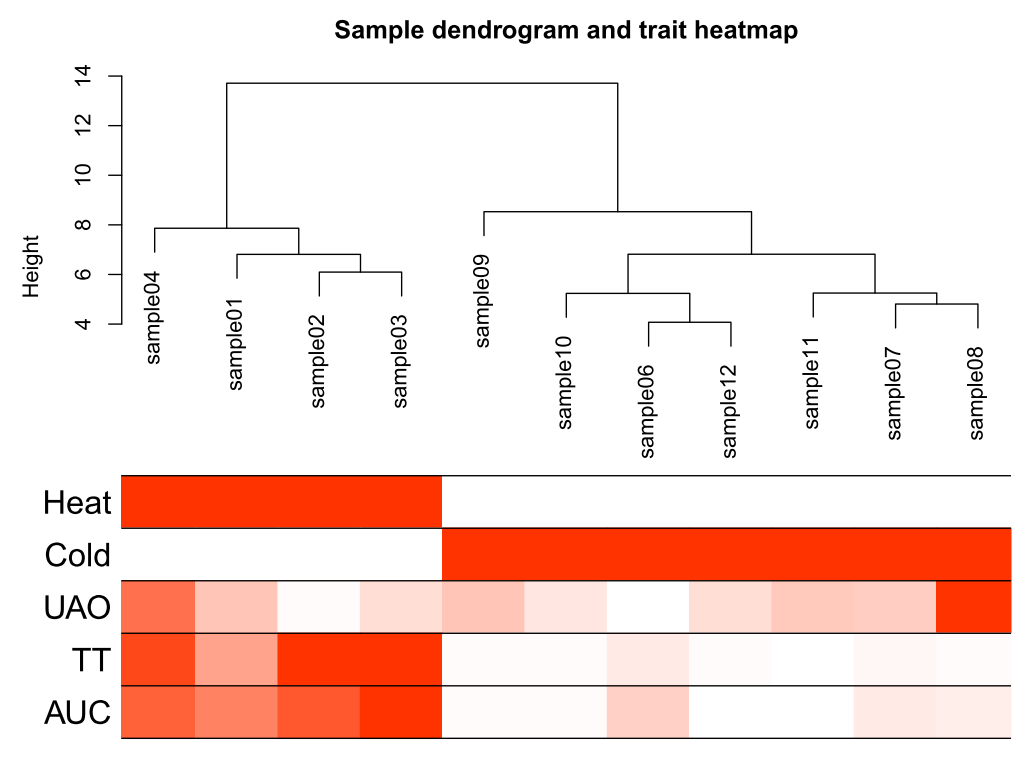
再次样本聚类
sampleTree2 = hclust(dist(datExpr0), method = "average")
# Convert traits to a color representation: white means low, red means high, grey means missing entry
traitColors = numbers2colors(datTraits, signed = FALSE)
输出样本聚类图
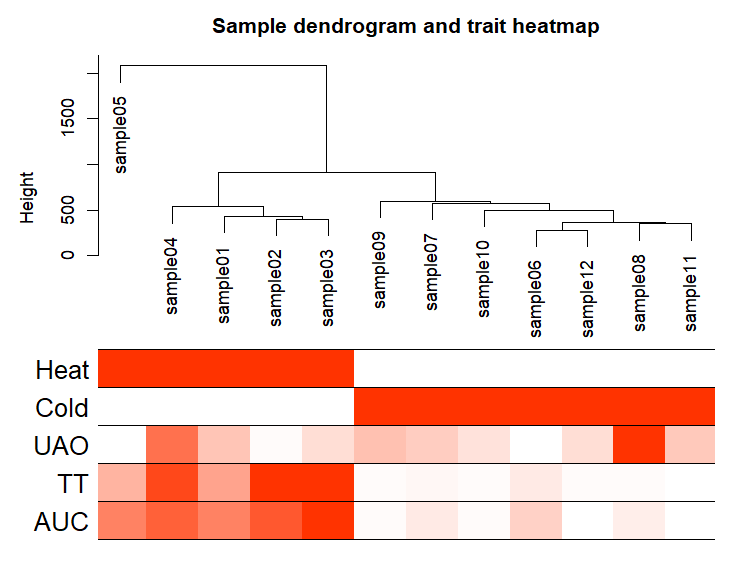
pdf(file="3_Sample_dendrogram_and_trait_heatmap.pdf",width=8 ,height= 6)
plotDendroAndColors(sampleTree2, traitColors,groupLabels = names(datTraits),main = "Sample dendrogram and trait heatmap",cex.colorLabels = 1.5, cex.dendroLabels = 1, cex.rowText = 2)
dev.off()
三、WGCNA分析(后面都是重点)
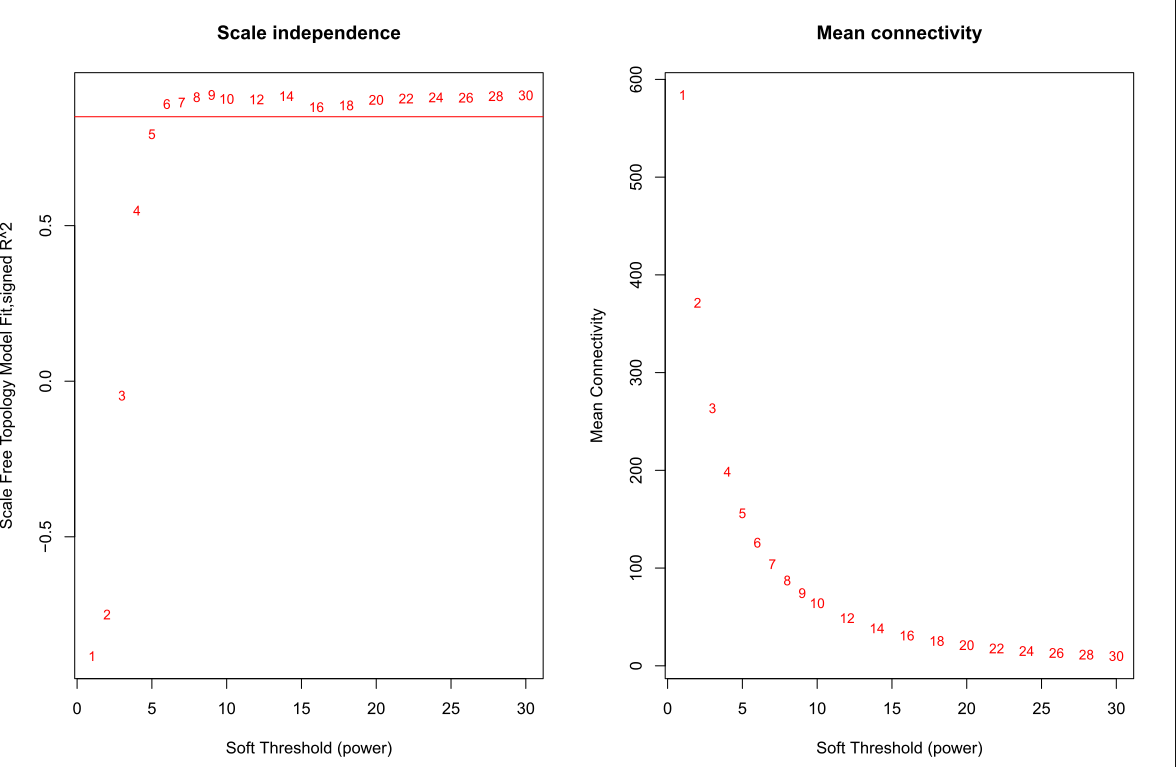
筛选软阈值
enableWGCNAThreads()
# 设置soft-thresholding powers的数量
powers = c(1:30)
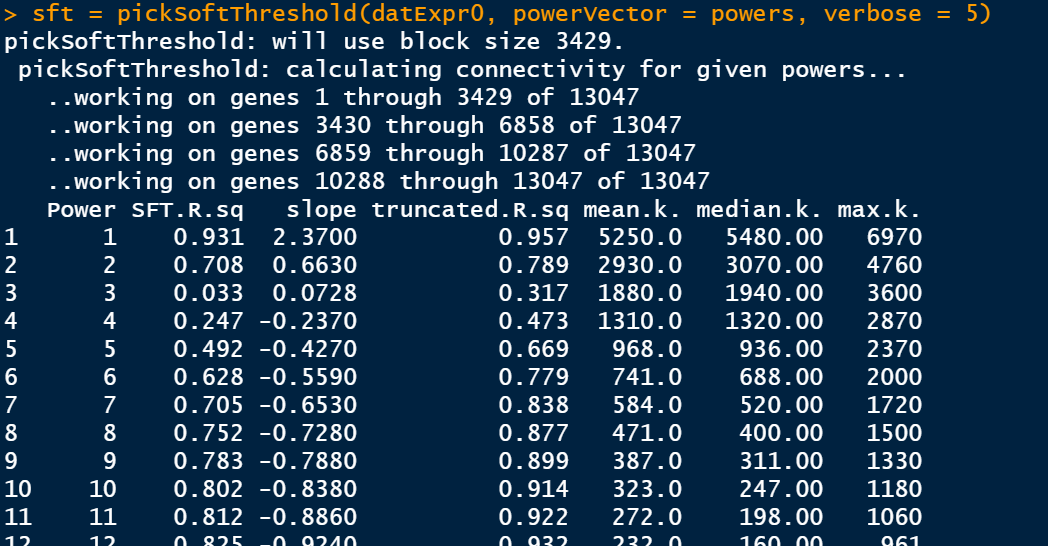
sft = pickSoftThreshold(datExpr0, powerVector = powers, verbose = 5)
此步骤是比较耗费时间的,静静等待即可。
绘制soft Threshold plot
#'@绘制soft power plot
pdf(file="4_软阈值选择.pdf",width=12, height = 8)
par(mfrow = c(1,2))
cex1 = 0.85
# Scale-free topology fit index as a function of the soft-thresholding power
plot(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2],xlab="Soft Threshold (power)",ylab="Scale Free Topology Model Fit,signed R^2",type="n",main = paste("Scale independence"));
text(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2],labels=powers,cex=cex1,col="red");
# this line corresponds to using an R^2 cut-off of h
abline(h=0.85,col="red")
# Mean connectivity as a function of the soft-thresholding power
plot(sft$fitIndices[,1], sft$fitIndices[,5],xlab="Soft Threshold (power)",ylab="Mean Connectivity", type="n",main = paste("Mean connectivity"))
text(sft$fitIndices[,1], sft$fitIndices[,5], labels=powers, cex=cex1,col="red")
dev.off()

选择softpower
选择softpower是一个玄学的过程,可以直接使用软件自己认为是最好的softpower值,但是不一定你要获得最好结果;其次,我们自己选择自己认为比较好的softpower值,但是,需要自己不断的筛选。因此,从这里开始WGCNA的分析结果就开始受到不同的影响。
## 选择软件认为是最好的softpower值
#softPower =sft$powerEstimate
---
# 自己设定softpower值
softPower = 9
继续分析
adjacency = adjacency(datExpr, power = softPower)
将邻接转化为拓扑重叠
这一步建议去服务器上跑,后面的步骤就在服务器上跑吧,数据量太大;如果你的数据量较小,本地也就可以
TOM = TOMsimilarity(adjacency);
dissTOM = 1-TOM
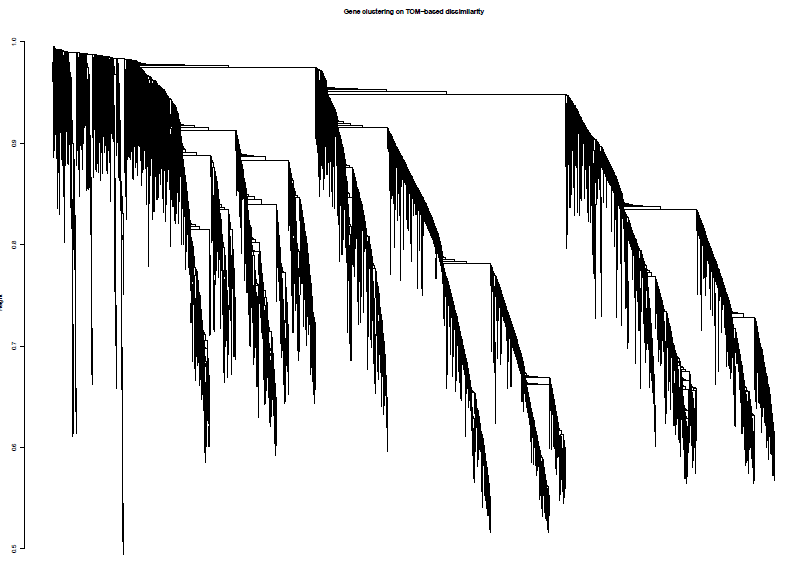
geneTree = hclust(as.dist(dissTOM), method = "average");
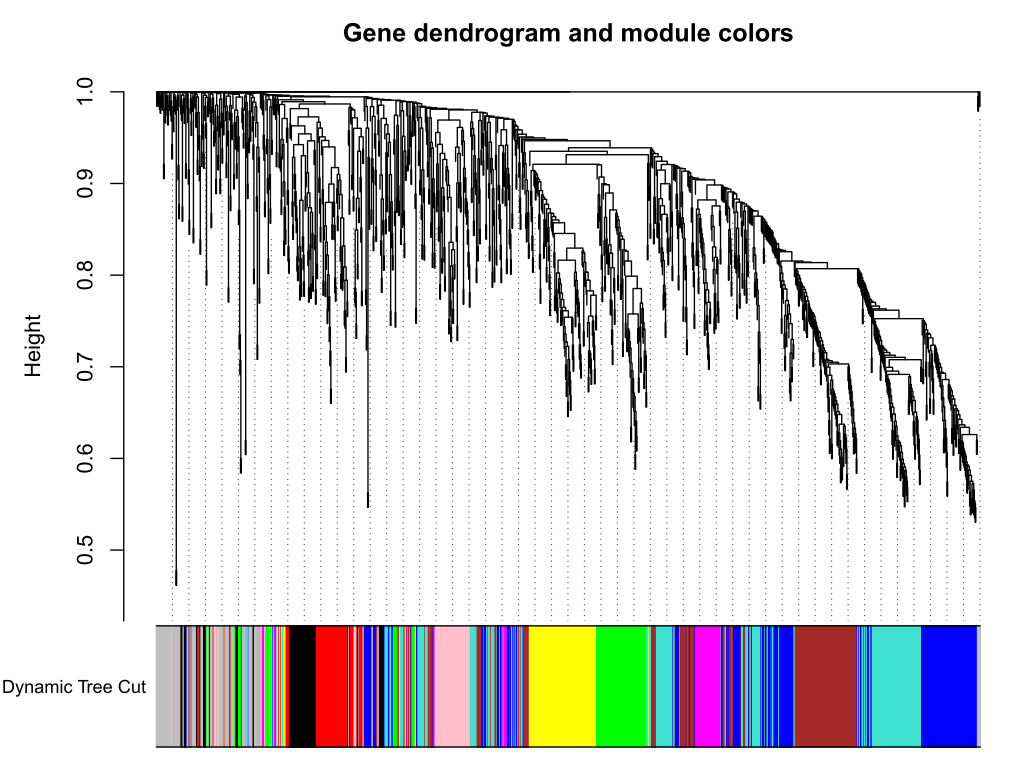
绘制聚类树(树状图)
pdf(file="4_Gene clustering on TOM-based dissimilarity.pdf",width=6,height=4)
plot(geneTree, xlab="", sub="", main = "Gene clustering on TOM-based dissimilarity",labels = FALSE, hang = 0.04)
dev.off()
加入模块
minModuleSize = 30
# Module identification using dynamic tree cut:
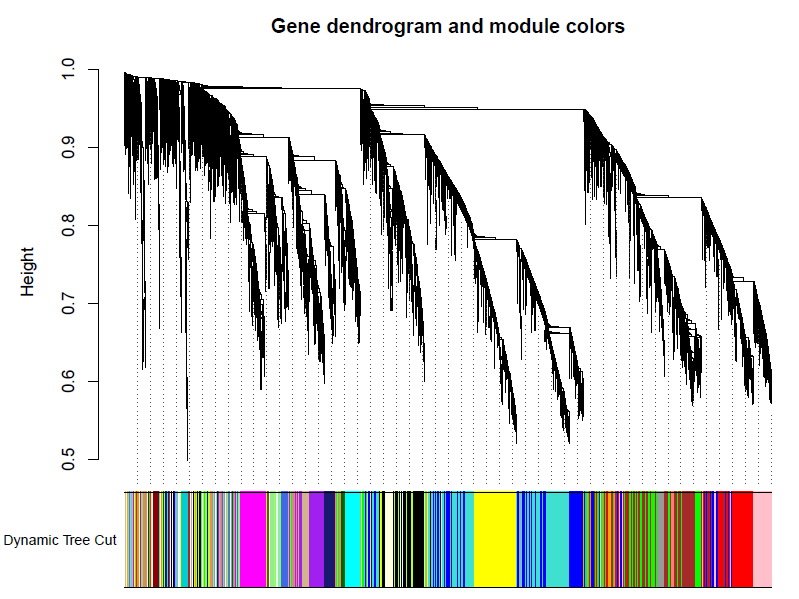
dynamicMods = cutreeDynamic(dendro = geneTree, distM = dissTOM,deepSplit = 2, pamRespectsDendro = FALSE,minClusterSize = minModuleSize);
table(dynamicMods)# Convert numeric lables into colors
dynamicColors = labels2colors(dynamicMods)
table(dynamicColors)
# Plot the dendrogram and colors underneath
#sizeGrWindow(8,6)
pdf(file="5_Dynamic Tree Cut.pdf",width=8,height=6)
plotDendroAndColors(geneTree, dynamicColors, "Dynamic Tree Cut",dendroLabels = FALSE, hang = 0.03,addGuide = TRUE, guideHang = 0.05,main = "Gene dendrogram and module colors")
dev.off()
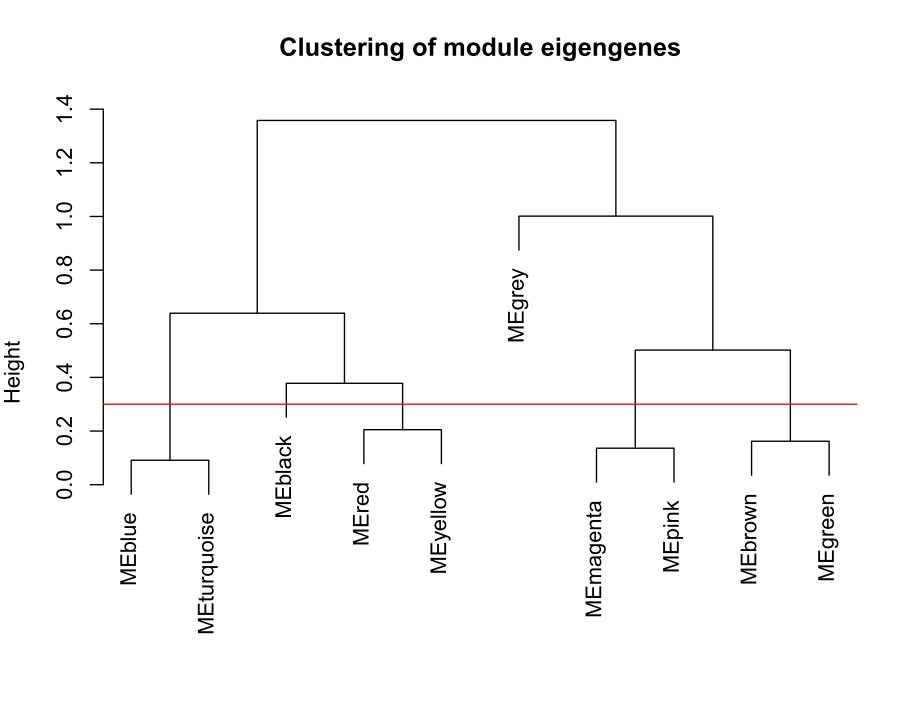
合并模块
做出的WGCNA分析中,具有较多的模块,但是在我们后续的分析中,是使用不到这么多的模块,以及模块越多对我们的分析越困难,那么就必须合并模块信息。具体操作如下。
MEList = moduleEigengenes(datExpr, colors = dynamicColors)
MEs = MEList$eigengenes
# Calculate dissimilarity of module eigengenes
MEDiss = 1-cor(MEs);
# Cluster module eigengenes
METree = hclust(as.dist(MEDiss), method = "average")
# Plot the result
#sizeGrWindow(7, 6)
pdf(file="6_Clustering of module eigengenes.pdf",width=7,height=6)
plot(METree, main = "Clustering of module eigengenes",xlab = "", sub = "")
MEDissThres = 0.3######剪切高度可修改
# Plot the cut line into the dendrogram
abline(h=MEDissThres, col = "red")
dev.off()

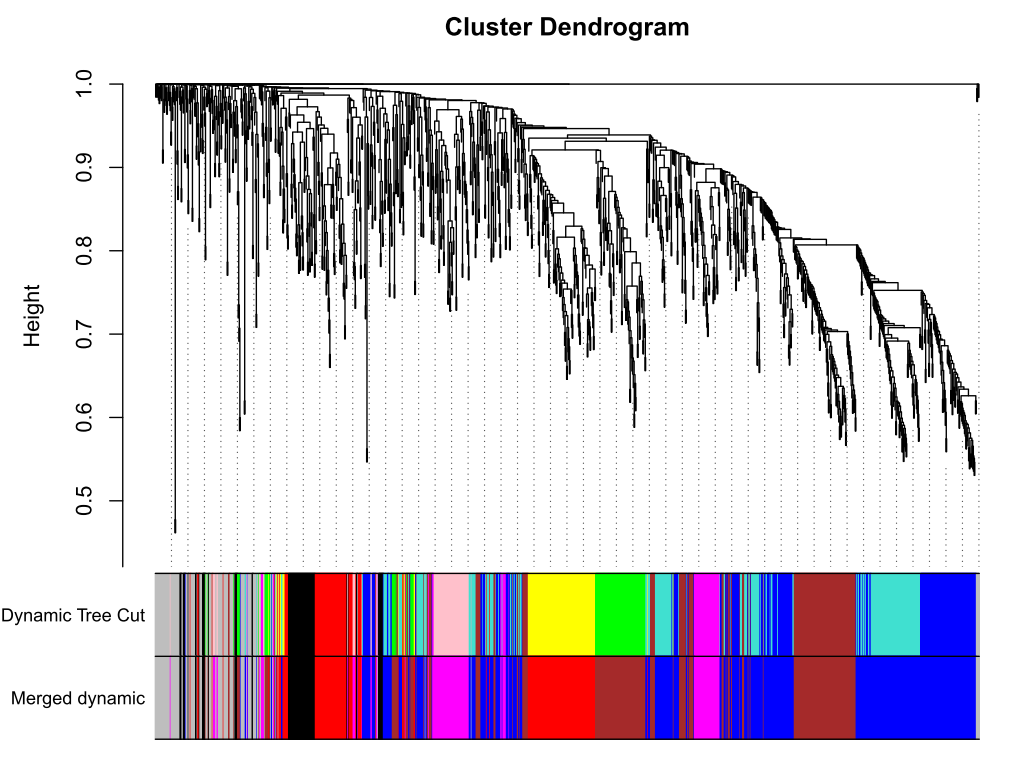
合并及绘图
merge = mergeCloseModules(datExpr, dynamicColors, cutHeight = MEDissThres, verbose = 3)
# The merged module colors
mergedColors = merge$colors
# Eigengenes of the new merged modules:
mergedMEs = merge$newMEs
table(mergedColors)#输出所有modules
color<-unique(mergedColors)
for (i in 1:length(color)) {y=t(assign(paste(color[i],"expr",sep = "."),datExpr[mergedColors==color[i]]))write.csv(y,paste('6',color[i],"csv",sep = "."),quote = F)
}
#save.image(file = "module_splitted.RData")
##'@输出merge模块图形
pdf(file="7_merged dynamic.pdf", width = 8, height = 6)
plotDendroAndColors(geneTree, cbind(dynamicColors, mergedColors),c("Dynamic Tree Cut", "Merged dynamic"),dendroLabels = FALSE, hang = 0.03,addGuide = TRUE, guideHang = 0.05)
dev.off()
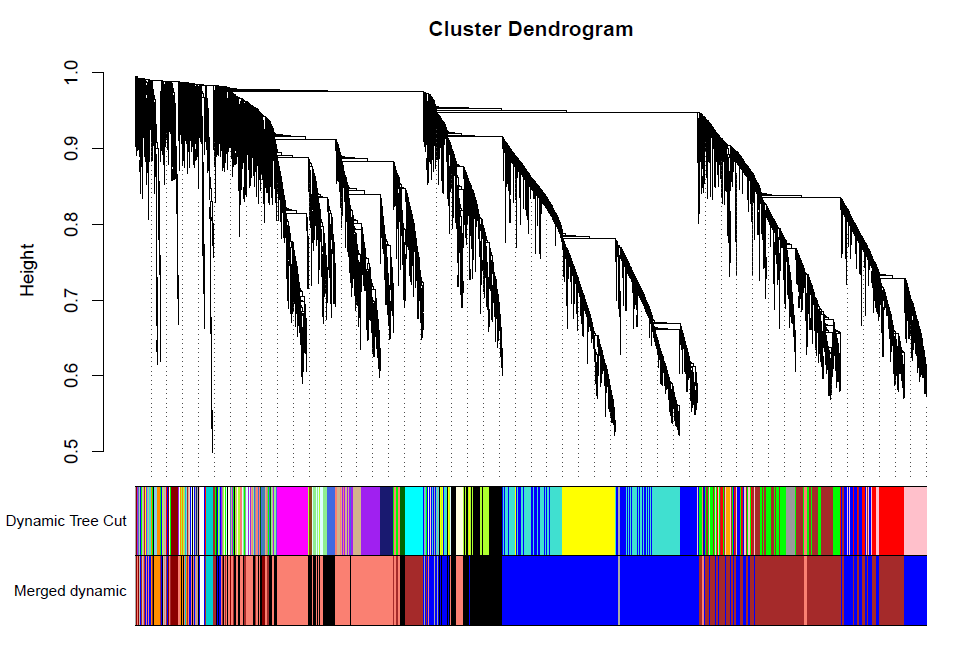
计算相关性
moduleColors = mergedColors
# Construct numerical labels corresponding to the colors
colorOrder = c("grey", standardColors(50))
moduleLabels = match(moduleColors, colorOrder)-1
MEs = mergedMEs
教程详情请看
https://mp.weixin.qq.com/s/ca8_eqirjJYgC8hiaF8jfg
往期文章:
1. 复现SCI文章系列专栏
2. 《生信知识库订阅须知》,同步更新,易于搜索与管理。

3. 最全WGCNA教程(替换数据即可出全部结果与图形)
-
WGCNA分析 | 全流程分析代码 | 代码一
-
WGCNA分析 | 全流程分析代码 | 代码二
-
WGCNA分析 | 全流程代码分享 | 代码三
-
WGCNA分析 | 全流程分析代码 | 代码四
4. 精美图形绘制教程
- 精美图形绘制教程
5. 转录组分析教程
转录组上游分析教程[零基础]
小杜的生信筆記 ,主要发表或收录生物信息学的教程,以及基于R的分析和可视化(包括数据分析,图形绘制等);分享感兴趣的文献和学习资料!!
这篇关于WGCNA分析教程五 | [更新版]的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!







