本文主要是介绍QIIME 2教程. 06沙漠土壤分析AtacamaSoil(2023.5),希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
QIIME 2用户文档. 6阿塔卡马沙漠微生物组分析

原文地址:https://docs.qiime2.org/2023.5/tutorials/atacama-soils/
此实例需要一些基础知识,要求完成本系列文章前两篇内容:《1简介和安装Introduction&Install》和《4人体各部位微生物组分析Moving Pictures》。
本教程设计用于两个目的。首先,它描述了对双端序列分析的初始处理步骤,直到分析步骤与单端序列分析相同。这包括导入、样本拆分和去噪步骤,并产生特征表和相关的特征序列。其次,这是一次自我练习,可以在《4人体各部位微生物组分析Moving Pictures》之后运行,以获得更多使用QIIME 2的经验。对于这个练习,我们提供了一些可以用来指导分析的问题,但是不提供直接解决每个问题的命令。相反,您应该应用您在《4人体各部位微生物组分析Moving Pictures》中学到的命令。
在本教程中,您将使用QIIME 2对来自智利北部阿塔卡马沙漠的土壤样本进行分析。阿塔卡马沙漠是地球上最干旱的地方之一,有些地区每十年降雨量不到一毫米。尽管极端干旱,土壤中仍然有微生物。本研究采样地点为东部的巴克达诺(Baquedano)和西部的永盖(Yungay),横断面的平均土壤相对湿度与海拔高度呈正相关(海拔越高,干旱程度越轻,平均土壤相对湿度越高)。沿着这些剖面,在每个地点挖坑,从每个坑的三个深度收集土壤样品。详见原文解读:mSystems:干旱对土壤微生物组的影响。
本节视频视频教程
https://www.bilibili.com/video/BV1DV411G7hg/
视频有广告,清晰度不够高吗?在公众号“宏基因组”后台回复“qiime2”获得1080p视频和测试数据下载链接。
启动QIIME2运行环境
对于上文提到了conda/docker两种常用安装方法,我们每次在分析数据前,需要打开工作环境,根据情况选择对应的打开方式。
# 定义工作目录变量,方便以后多次使用
wd=~/mnt/d/github/QIIME2ChineseManual/2023.5
mkdir -p $wd
# 进入工作目录,是不是很简介,这样无论你在什么位置就可以快速回到项目文件夹
cd $wd# 方法1. 进入QIIME 2 conda工作环境
conda activate qiime2-2023.5
# 这时我们的命令行前面出现 (qiime2-2023.5) 表示成功进入工作环境# 方法2. conda版本较老用户,使用source进入QIIME 2
source activate qiime2-2023.5# 方法3. 如果是docker安装的请运行如下命令,默认加载当前目录至/data目录
docker run --rm -v $(pwd):/data --name=qiime -it qiime2/core:2023.5# 创建本节学习目录
mkdir -p atacama
cd atacama实验数据下载
Obtain the data
注意:QIIME 2 官方测试数据部分保存在Google服务器上,国内下载比较困难。公众号后台回复”qiime2”获取测试数据批量下载链接,你还可以跳过以后的wget步骤。
下载Google文档的国内备份实验设计
http://www.imeta.science/github/QIIME2ChineseManual/2023.5/atacama/sample-metadata.tsv下载双端实验数据(使用10%抽样数据方便下载和演示):分别为正向、反向和barcodes序列三个文件;文来自亚马逊云,有时无法下载或断开,可不同时间多试几次就成功了。或使用后台百度云链接,或github备份永久链接。
注:github有些文件过大无法上传。建议自行原始地址下载。或后台百度云链接下载(可能会失效)。
# mkdir创建序列存放目录
# -p 参数创建目录,即使目录存在也不报错
mkdir -p emp-paired-end-sequences# wget下载文件
# -c为支持断点续传,跨国下载经常断,-c必须加,否则你下不完断线又要从头下载
wget -c \-O "emp-paired-end-sequences/forward.fastq.gz" \"https://data.qiime2.org/2023.5/tutorials/atacama-soils/10p/forward.fastq.gz"
wget -c \-O "emp-paired-end-sequences/reverse.fastq.gz" \"https://data.qiime2.org/2023.5/tutorials/atacama-soils/10p/reverse.fastq.gz"
wget -c \-O "emp-paired-end-sequences/barcodes.fastq.gz" \"https://data.qiime2.org/2023.5/tutorials/atacama-soils/10p/barcodes.fastq.gz"双端数据分析方法
Paired-end read analysis commands
双端数据导入,数据建库类型为EMP双端序列EMPPairedEndSequences(本示例来自EMP项目)
# 笔记本:23s,服务器34s
time qiime tools import \--type EMPPairedEndSequences \--input-path emp-paired-end-sequences \--output-path emp-paired-end-sequences.qza导入的压缩文件有300 MB,约耗时1m。
输出对象:
emp-paired-end-sequences.qza: EMP项目双端测序类型。查看 | 下载
接下来,我们按Barcode序列信息进行样品拆分。这需要样本元数据文件,您必须指明该文件中的哪个列包含每个样本的条形码。在这种情况下,该列名称是条形码序列barcode-sequence。在此数据集中,条形码读长是样本元数据文件中包含的条形码读长的反向互补序列,因此我们还包括--p-rev-comp-mapping-barcodes参数。在样品拆分之后,我们可以生成并查看每个样本获得多少序列的摘要。
# 样本拆分,EMP双端序列类型,指定元数据,barcode所在列,
# 并序列取反向互补(barcode加有右端时使用),i为输出文件,o为输出文件和统计
time qiime demux emp-paired \--m-barcodes-file sample-metadata.tsv \--m-barcodes-column barcode-sequence \--p-rev-comp-mapping-barcodes \--i-seqs emp-paired-end-sequences.qza \--o-per-sample-sequences demux-full.qza \--o-error-correction-details demux-details.qza
# 笔记本:2m3s,服务器:3m4s输出对象:
demux-full.qza: 样品拆分结果文件。查看 | 下载
demux-details.qza:样品拆分统计文件。
查看 | 下载
结果可视化:
qiime demux summarize \--i-data demux-full.qza \--o-visualization demux-full.qzv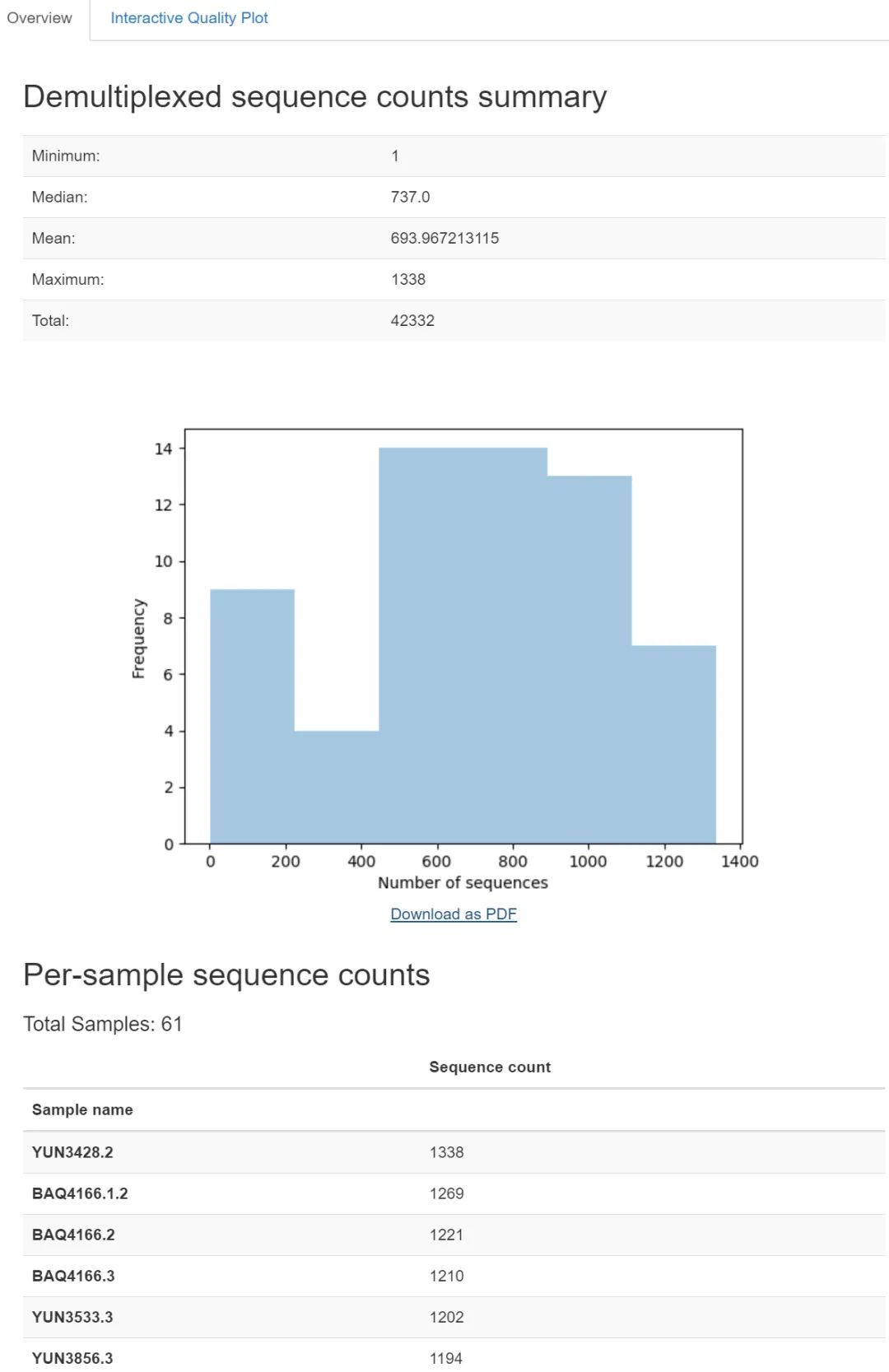
图1. 数据量汇总图表。中位数有737,可以分析练手了。
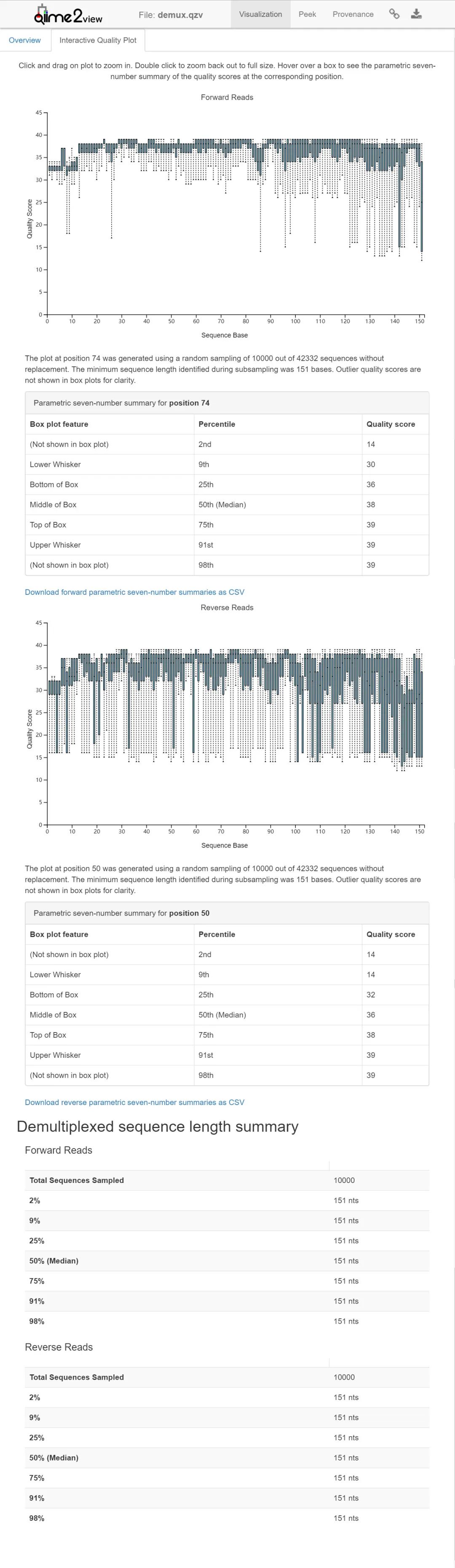
图2. 双端数据质量评估图。
让我们对数据进行重采样。我们将在本教程中执行此重采样有两个原因:一个是为了加快教程的运行时间,另一个是为了演示功能。
注意,下面的重采样示例旨在说明q2-demux的重采样功能。如果您正在考虑对序列进行重抽样,请确保您已仔细考虑并有合理理由。
qiime demux subsample-paired \--i-sequences demux-full.qza \--p-fraction 0.3 \--o-subsampled-sequences demux-subsample.qzaqiime demux summarize \--i-data demux-subsample.qza \--o-visualization demux-subsample.qzv输出对象:
demux-subsample.qza:拆分-重采样文件。
查看 | 下载
输出可视化:
demux-subsample.qzv:拆分-重样可视化。
查看 | 下载
让我们看看demux-subsample.qzv中的概况。在“Overview”选项卡上的“按样本序列计数”表中,数据中有75个样本。不过,如果我们查看表中的最后20行左右,我们会发现许多样本中的序列数少于100——让我们从数据中过滤掉这些样本。
注意,以下过滤样本的示例旨在说明q2-demux的过滤能力,如果您考虑从研究中过滤样本,请确保您已仔细考虑并具有合理的理由。
qiime tools export \--input-path demux-subsample.qzv \--output-path ./demux-subsample/qiime demux filter-samples \--i-demux demux-subsample.qza \--m-metadata-file ./demux-subsample/per-sample-fastq-counts.tsv \--p-where 'CAST([forward sequence count] AS INT) > 100' \--o-filtered-demux demux.qza输出对象:
demux.qza:demux的输出对象。
查看 | 下载
去噪并生成特征表和代表序列
接下来,我们将来看看这些序列的质量,这些序列是从过滤后的数据中经过10000次随机重采样产生的,然后对数据进行去噪。当你查看质量图时,请注意,与moving pictures教程中的相应图相比,现在有两个交互式图要一起考虑。左侧的图显示了正向序列的质量得分,右侧的图显示了反向序列的质量得分。我们将使用这些图来确定要用于DADA2去噪的调整参数,然后使用dada2 denoise-paired进行去噪。
在此示例中,我们有150个碱基的正向和反向序列。由于我们需要序列的长度足够长,以便在合并序列末端时重叠,因此正向和反向序列的前13个碱基将被修剪,但不会对序列的末端进行修剪,以免修剪太长的序列。在此示例中,为--p-trim-left-f和--p-trim-left-r以及--p-trunc-len-f和--p-trunc-len-r提供了相同修剪数值,但这不是必须的。
# 笔记本:2m36s,服务器:4m20s
time qiime dada2 denoise-paired \--i-demultiplexed-seqs demux.qza \--p-trim-left-f 13 \--p-trim-left-r 13 \--p-trunc-len-f 150 \--p-trunc-len-r 150 \--o-table table.qza \--o-representative-sequences rep-seqs.qza \--o-denoising-stats denoising-stats.qza输出对象:
denoising-stats.qza:去噪结果统计。
查看 | 下载
rep-seqs.qza:代表序列。
查看 | 下载
table.qza:表格。
查看 | 下载
在这一阶段,您将拥有包含特征表、相应特征序列和DADA2去噪统计信息的对象。您可以生成这些摘要。
qiime feature-table summarize \--i-table table.qza \--o-visualization table.qzv \--m-sample-metadata-file sample-metadata.tsvqiime feature-table tabulate-seqs \--i-data rep-seqs.qza \--o-visualization rep-seqs.qzvqiime metadata tabulate \--m-input-file denoising-stats.qza \--o-visualization denoising-stats.qzv可视化输出结果:
table.qzv:表格。
查看 | 下载
denoising-stats.qzv:去噪统计。
查看 | 下载
rep-seqs.qzv代表序列:。
查看 | 下载
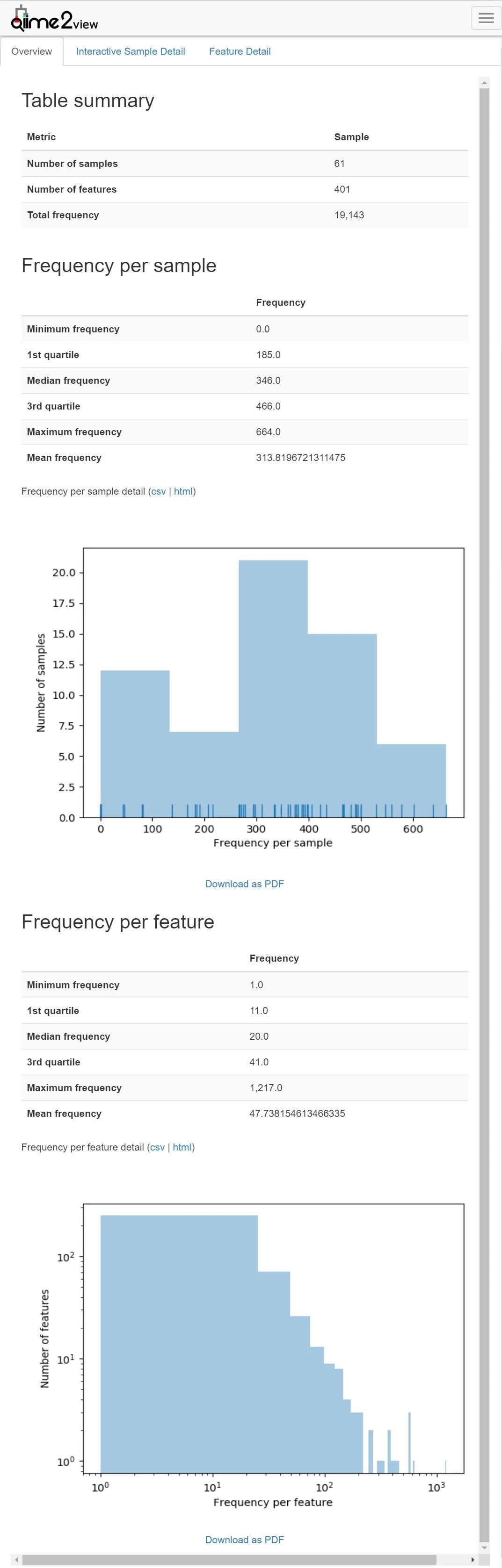
图3. 特征表统计
我们要根据数据量,来选择合适的重采样值
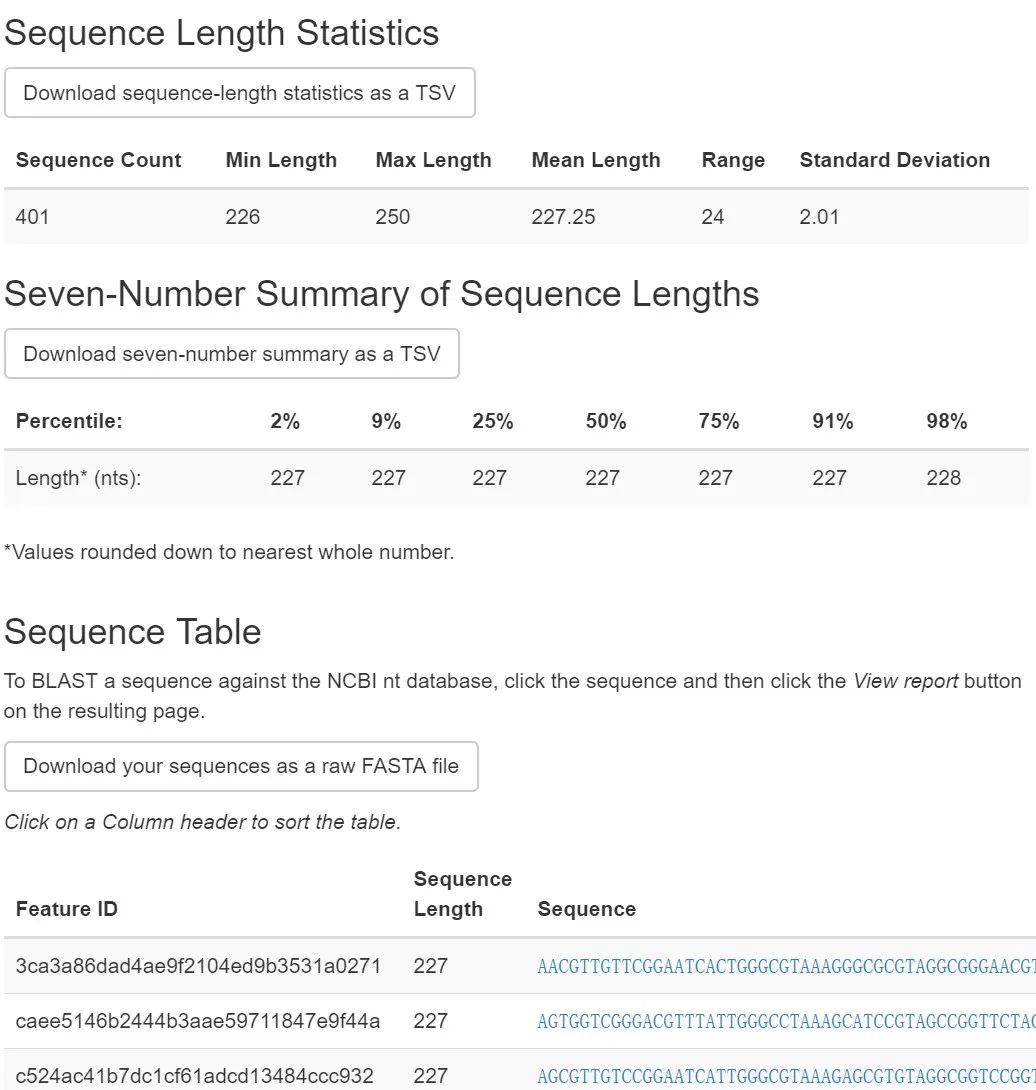
图4. 代表性序列统计
长度基本全一致,意义不大。可以点击序列查询相关注意比较方便。
从这个地方开始,双端序列和单端序列的分析方法就相同了。因此,你可以参照《4、人体各部位微生物组分析Moving Picture(2020.2版)》教程中的方法继续分析你接下来的步骤。
接下来分析要回答的科学问题
Questions to guide data analysis
通过以下问题,来指导你分析数据。
接下来特征表重采样标准化参数
--p-sampling-depth应该选多少?基于你重采样的参数,有多少样品应该从实验中剔除?
在
core-metrics-phylogenetic分析中,使用过滤后的样本有多少数据量?实验设计中的那种分组方式下微生物组成差异最大?
采用那种距离计算方法分开更明显,是
unweighted UniFrac还是Bray-Curtis?根据你对这些距离计算方法的理解,这些不同代表什么意义呢?
对于连续型的样本属性,考虑尝试使用
qiime metadata distance-matrix与qiime diversity mantel和qiime diversity bioenv结合使用更有效,这些命令之前没有提到过,但可以使用--help查看详细帮助。分析样本连续型属性与样本的丰富多、均匀度之间的关系?
推荐使用
qiime diversity alpha-correlation分析多样性与样本属性间的相关性,看看能得到什么结论?不会记得查看帮助文档。
哪种样本的分类与Alpha多样性差异最相关,并比较是否有显著差异?
在门水平查看不同土壤相对温度下微生物组成,哪个门丰度最高?
看那些种类与湿度正/负相关?
在有无植被的取样地点,什么菌门差异明显?
参考答案
重抽样数量的选择
接下来特征表重采样标准化参数
--p-sampling-depth应该选多少?基于你重采样的参数,有多少样品应该从实验中剔除?
在
core-metrics-phylogenetic分析中,使用过滤后的样本有多少数据量?
想要选择合适的重采样参数,需要使用 https://view.qiime2.org/ 查看 table.qzv 文件,首页即有样本的数据量分布。我们看到有大量样本测序量仅为1000左右。精确选择,要切换至Interactive Sample Detail面板,下拉看最下面的每个样本量排序的表格。有大量的1000可以直接去掉,这里可以选1446保留尽可能多的样品;也可选3041,只去掉5个3千以下的样本。此处先选大值,因为分析中可以尽量扔样本,越高质量结果越少越容易发现规律,不够再找。我的也不是标准答案,仅供参数,分析中结果是否最优是尝试出来的。我在此处并没有太多尝试,即有经验,也有个人的习惯。
进化树构建和多样性分析
# 15s/23s
time qiime phylogeny align-to-tree-mafft-fasttree \--i-sequences rep-seqs.qza \--o-alignment aligned-rep-seqs.qza \--o-masked-alignment masked-aligned-rep-seqs.qza \--o-tree unrooted-tree.qza \--o-rooted-tree rooted-tree.qza# 12s,采样深度来自table.qzv,最小值仅有24,取Q1=931
time qiime diversity core-metrics-phylogenetic \--i-phylogeny rooted-tree.qza \--i-table table.qza \--p-sampling-depth 931 \--m-metadata-file sample-metadata.tsv \--output-dir core-metrics-results环境因子关联分析
实验设计中的那种分组方式下微生物组成差异最大?
采用那种距离计算方法分开更明显,是
unweighted UniFrac还是Bray-Curtis?根据你对这些距离计算方法的理解,这些不同代表什么意义呢?
对于连续型的样本属性,考虑尝试使用
qiime metadata distance-matrix与qiime diversity mantel和qiime diversity bioenv结合使用更有效,这些命令之前没有提到过,但可以使用--help查看详细帮助。
我们可以先查看各种距离的 qzv 中切换不同的分组方式查看,这里仅选择 bray(bray_curtis_emperor.qzv)和unifrac(weighted_unifrac_emperor.qzv)两种演示。在Color中选择不同分类属性,如ph、humidity等属性,其中relative-humidity-soil-low中不同颜色在PCoA的前三轴分开明显。同时要注意每个轴上的解析率。尝试不同的方法,如unifrac中各轴解析率会比bray方法下高很多,是什么原因呢? 可以查查这两种方法的计算原理就清楚了。
接下来我们探索几种可以统计连续型变量的统计方法。qiime metadata distance-matrix与qiime diversity mantel和qiime diversity bioenv
首先学习—help查看metadata distance-matrix命令的帮助,看一下新命令的介绍
qiime metadata distance-matrix --helpUsage: qiime metadata distance-matrix [OPTIONS]Create a distance matrix from a numeric metadata column. The Euclideandistance is computed between each pair of samples or features in thecolumn.Tip: the distance matrix produced by this method can be used as input tothe Mantel test available in `q2-diversity`.Parameters:--m-metadata-file METADATA--m-metadata-column COLUMN MetadataColumn[Numeric]Numeric metadata column to compute pairwise Euclideandistances from [required]
Outputs:--o-distance-matrix ARTIFACTDistanceMatrix [required]
Miscellaneous:--output-dir PATH Output unspecified results to a directory--verbose / --quiet Display verbose output to stdout and/or stderr duringexecution of this action. Or silence output ifexecution is successful (silence is golden).--citations Show citations and exit.--help Show this message and exit.它是计算元数据/特征中连续数值变量间欧式距离矩阵中命令。输入文件为元数据和列名。输出为qiime2对象qza
time qiime metadata distance-matrix \--m-metadata-file sample-metadata.tsv \--m-metadata-column relative-humidity-soil-low \--o-distance-matrix sample-metadata-relative-humidity-soil-low.qzaPlugin error from metadata:Encountered missing value(s) in the metadata column. Computing a distance matrix from missing values is not supported. IDs with missing values: BAQ4697.1, BAQ4697.2, BAQ4697.3错误提示:数据中存在错误值,无法计算。一般需要手动在实验设计中移除缺失的样本,再计算。
我们计算一个信息完整的海拔列
qiime metadata distance-matrix \--m-metadata-file sample-metadata.tsv \--m-metadata-column elevation \--o-distance-matrix sample-metadata-elevation.qzaqiime diversity mantel可以基于特征的距离矩阵,和样本元数据的距离矩阵,计算两者间的相关性。找到和微生物群落结构变化的相关因素。
qiime diversity mantel \--i-dm1 core-metrics-results/weighted_unifrac_distance_matrix.qza \--i-dm2 sample-metadata-elevation.qza \--p-method spearman \--p-intersect-ids True \--p-label1 weighted_unifrac \--p-label2 elevation \--o-visualization core-metrics-results/weighted_unifrac_evaluation.qzv我们看到 海拔 与 unifrac间存在显著相关。
qiime diversity bioenv计算元数据的欧式距离中那一类与距离矩阵秩最大相关。其中所有的数字列都会考虑,缺失值会自动移除,输出可视化结果。
# 原始数据中列太多,也存在错误;只提取其中部分
cut -f 1-5,10,13 sample-metadata.tsv > temp
# 计算数字列的相关性
time qiime diversity bioenv \--i-distance-matrix core-metrics-results/weighted_unifrac_distance_matrix.qza \--m-metadata-file temp \--o-visualization core-metrics-results/weighted-unifrac-bioenv.qzvaverage-soil-relative-humidity是最大相关因素。
alpha多样性与连续性变量分析
分析样本连续型属性与样本的丰富多、均匀度之间的关系?
推荐使用
qiime diversity alpha-correlation分析多样性与样本属性间的相关性,看看能得到什么结论?不会记得查看帮助文档。
我们以observed_otus(richness)为例
# 查看帮助
qiime diversity alpha-correlation --helpqiime diversity alpha-correlation \--i-alpha-diversity core-metrics-results/observed_features_vector.qza \--m-metadata-file sample-metadata.tsv \--o-visualization core-metrics-results/observed_features_correlation.qzv看到elevation与richness显著相关,再Column切换其它参数,如average-soil-relative-humidity相关性更好,高达0.6909。
哪种样本的分类与Alpha多样性差异最相关,并比较是否有显著差异?
同上一题。查看core-metrics-results/observed_otus_correlation.qzv结果即有答案。
物种组成差异及相关分析
在门水平查看不同土壤相对温度下微生物组成,哪个门丰度最高?
看那些种类与湿度正/负相关?
想分析门水平,必须先物种注释,再统计组成。
# 物种注释和可视化,使用上一节下载的数据库
# 48s,1m24s
time qiime feature-classifier classify-sklearn \--i-classifier ../moving-pictures/gg-13-8-99-515-806-nb-classifier.qza \--i-reads rep-seqs.qza \--o-classification taxonomy.qza# 生成物种可视化,即每个Feature对应的物种注释和可信度
qiime metadata tabulate \--m-input-file taxonomy.qza \--o-visualization taxonomy.qzv# 物种组成柱状图,按Level2和样本类型 sample-type 排序
qiime taxa barplot \--i-table table.qza \--i-taxonomy taxonomy.qza \--m-metadata-file sample-metadata.tsv \--o-visualization taxa-bar-plots.qzv查看taxa-bar-plots.qzv,按Taxonomic Level选择Level 2,Sort按average-soil-temperature,可以查看放线菌丰度最高。相关性分析如何呢?大家想一想。
在有无植被的取样地点,什么菌门差异明显?
# 按属比较,需要先合并
qiime taxa collapse \--i-table table.qza \--i-taxonomy taxonomy.qza \--p-level 2 \--o-collapsed-table table-l2.qza# 格式转换
qiime composition add-pseudocount \--i-table table-l2.qza \--o-composition-table comp-table-l2.qza# 按有无植被差异比较
time qiime composition ancom \--i-table comp-table-l2.qza \--m-metadata-file sample-metadata.tsv \--m-metadata-column vegetation \--o-visualization l6-ancom-vegetation.qzv
# 分类学差异直接有名称,不用feature再对应物种注释译者简介
刘永鑫,研究员,博士生导师。2014年博士毕业于中国科学院大学生物信息学专业,之后在中国科学院遗传与发育生物学研究所工作历任博士后、工程师、高级工程师,2022年10月加入中国农业科学院深圳农业基因组研究所担任课题组长。研究方向为宏基因组方法开发、功能挖掘和科学传播。参与QIIME 2项目,主导开发了易扩增子(EasyAmplicon)、易宏基因组(EasyMetagenome)、培养组(Culturome)分析流程、数据分析网站(EVenn、ImageGP) 和R包(amplicon、ggClusterNet)等,目标是全面打造宏基因组领域方法学基础设施,推动微生物组学发展。以(共同)第一或通讯作者在Nature Biotechnology、Nature Microbiology、iMeta等期刊发表论文20余篇。合作在Science、Cell Host & Microbe、Microbiome等期刊发表论文20余篇,累计发表论文50余篇,被引用13000+次。主编《微生物组实验手册》专著,由300多位同行参与,共同打造本领域长期更新的中文百科全书。创办宏基因组公众号,15万+同行关注,分享原创文章3千余篇,累计阅读量超4千万,打造本领域最具影响力的科学传播平台。发起《iMeta》期刊,联合全球千位专家共同打造宏基因组学、微生物组和生物信息学顶刊,解决我国本领域期刊出版卡脖子问题。课题组长期招聘博士后、客座研究生,有兴趣可加微信yongxinliu详谈。
王惠铃,湖南农业大学,生物信息学本科在读,在刘永鑫组毕业实习。负责本次版本的更新和测试。
Reference
https://docs.qiime2.org/2023.5
Evan Bolyen, Jai Ram Rideout, Matthew R. Dillon, Nicholas A. Bokulich, Christian C. Abnet, Gabriel A. Al-Ghalith, Harriet Alexander, Eric J. Alm, Manimozhiyan Arumugam, Francesco Asnicar, Yang Bai, Jordan E. Bisanz, Kyle Bittinger, Asker Brejnrod, Colin J. Brislawn, C. Titus Brown, Benjamin J. Callahan, Andrés Mauricio Caraballo-Rodríguez, John Chase, Emily K. Cope, Ricardo Da Silva, Christian Diener, Pieter C. Dorrestein, Gavin M. Douglas, Daniel M. Durall, Claire Duvallet, Christian F. Edwardson, Madeleine Ernst, Mehrbod Estaki, Jennifer Fouquier, Julia M. Gauglitz, Sean M. Gibbons, Deanna L. Gibson, Antonio Gonzalez, Kestrel Gorlick, Jiarong Guo, Benjamin Hillmann, Susan Holmes, Hannes Holste, Curtis Huttenhower, Gavin A. Huttley, Stefan Janssen, Alan K. Jarmusch, Lingjing Jiang, Benjamin D. Kaehler, Kyo Bin Kang, Christopher R. Keefe, Paul Keim, Scott T. Kelley, Dan Knights, Irina Koester, Tomasz Kosciolek, Jorden Kreps, Morgan G. I. Langille, Joslynn Lee, Ruth Ley, Yong-Xin Liu, Erikka Loftfield, Catherine Lozupone, Massoud Maher, Clarisse Marotz, Bryan D. Martin, Daniel McDonald, Lauren J. McIver, Alexey V. Melnik, Jessica L. Metcalf, Sydney C. Morgan, Jamie T. Morton, Ahmad Turan Naimey, Jose A. Navas-Molina, Louis Felix Nothias, Stephanie B. Orchanian, Talima Pearson, Samuel L. Peoples, Daniel Petras, Mary Lai Preuss, Elmar Pruesse, Lasse Buur Rasmussen, Adam Rivers, Michael S. Robeson, Patrick Rosenthal, Nicola Segata, Michael Shaffer, Arron Shiffer, Rashmi Sinha, Se Jin Song, John R. Spear, Austin D. Swafford, Luke R. Thompson, Pedro J. Torres, Pauline Trinh, Anupriya Tripathi, Peter J. Turnbaugh, Sabah Ul-Hasan, Justin J. J. van der Hooft, Fernando Vargas, Yoshiki Vázquez-Baeza, Emily Vogtmann, Max von Hippel, William Walters, Yunhu Wan, Mingxun Wang, Jonathan Warren, Kyle C. Weber, Charles H. D. Williamson, Amy D. Willis, Zhenjiang Zech Xu, Jesse R. Zaneveld, Yilong Zhang, Qiyun Zhu, Rob Knight & J. Gregory Caporaso#. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nature Biotechnology. 2019, 37(8): 852-857. https://doi.org/10.1038/s41587-019-0209-9
The data used in this tutorial is presented in: Significant Impacts of Increasing Aridity on the Arid Soil Microbiome. Julia W. Neilson, Katy Califf, Cesar Cardona, Audrey Copeland, Will van Treuren, Karen L. Josephson, Rob Knight, Jack A. Gilbert, Jay Quade, J. Gregory Caporaso, and Raina M. Maier. mSystems May 2017, 2 (3) e00195-16; DOI: 10.1128/mSystems.00195-16.
猜你喜欢
iMeta简介 高引文章 高颜值绘图imageGP 网络分析iNAP
iMeta网页工具 代谢组MetOrigin 美吉云乳酸化预测DeepKla
iMeta综述 肠菌菌群 植物菌群 口腔菌群 蛋白质结构预测
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature
系列教程:微生物组入门 Biostar 微生物组 宏基因组
专业技能:学术图表 高分文章 生信宝典 不可或缺的人
一文读懂:宏基因组 寄生虫益处 进化树 必备技能:提问 搜索 Endnote
扩增子分析:图表解读 分析流程 统计绘图
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流快速解决科研困难,我们建立了“宏基因组”讨论群,己有国内外6000+ 科研人员加入。请添加主编微信meta-genomics带你入群,务必备注“姓名-单位-研究方向-职称/年级”。高级职称请注明身份,另有海内外微生物PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。
点击阅读原文,跳转最新文章目录阅读
这篇关于QIIME 2教程. 06沙漠土壤分析AtacamaSoil(2023.5)的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!








