本文主要是介绍单细胞数据分布 ZINB的理解,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
单细胞RNA测序(single-cell RNA-seq,scRNA-seq)数据是非常有特点的数据,具有很高的稀疏性(high sparsity),具体表现为0非常多(zero inflation)。对于数据的分布给出合理的假设是非常关键的工作,是downstream analysis的基础。显然对于scRNA-seq的reads count数据,最常用的正态分布是不合理的。首先正态分布描述的是连续型数据,而reads count数据是离散的;其次reads count数据的取值只能为非负整数。经过不断的尝试,ZINB被证明是一种可以较好的描述scRNA-seq数据的模型,并且作为一些更advanced的模型的基础比如SAVER,scVI等。下面我们来看这个模型的细节。
1 Poisson Distribution
基于reads count数据的取值均为非负整数的特点,一个直观的想法就是用泊松分布来拟合scRNA-seq数据。泊松分布的定义如下:
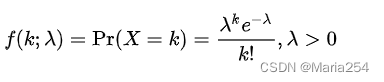
这里X即为gene在细胞内的表达水平(reads count的数值)。但是用泊松分布来描述scRNA-seq数据面临了一个新的问题。我们都知道,泊松分布的期望和方差是相等的,即:

但是对于实际的数据来说,随着gene的平均表达水平越高,其样本方差与样本均值的差越大,也即scRNA-seq数据的另一个特点——over-dispersion。我们用一张图来举例说明
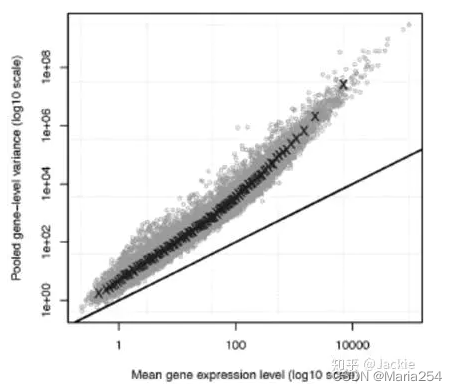
如图所示,直线(y = x)为基于泊松分布的假设下,基因表达的理论均值与方差的关系,可以看到对于每一种基因,其理论均值与方差相同。而直线之上的部分体现了实际数据中,基因表达的样本均值与样本方差的关系,我们看到,随着基因表达样本均值的增大,基因表达的样本方差与均值的差越来越大,不符合泊松分布的性质。
~~
2 Gamma Distribution
~~
对于泊松分布来说,是固定不变的,如果我们给一个prior呢。 我们关于prior的选择是Gamma分布。而选择Gamma分布作为的prior在生物学含义上似乎没有比较直观的解释(其实是我自己没搞懂hhh),但是从统计观点看,Gamma分布是泊松分布的共轭先验(conjugate prior),会使得计算posterior非常方便。
Gamma分布的定义如下:
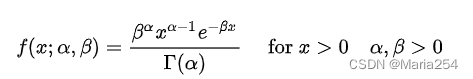
3 Negative Binomial Distribution
上述问题现在汇总为:
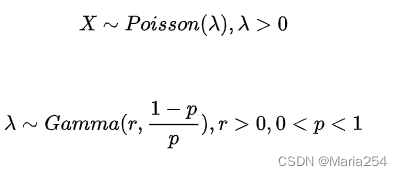
证明X服从负二项分布:
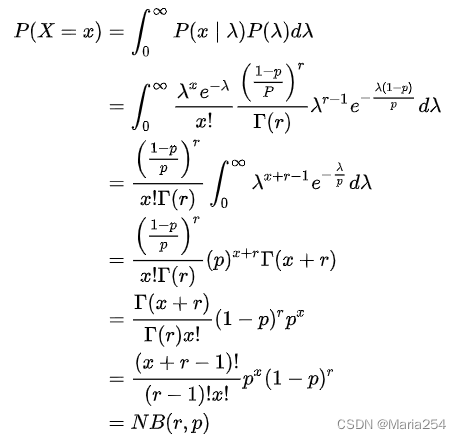
根据上述证明,X服从负二项分布。但是新的问题接着产生,在产生数据的过程中,由于一些technical noises(比如某段RNA没有能够被逆转录)和intrinsic biological variability会导致数据中0的比例非常高,这也就是所谓的zero inflation。于是人们在NB的基础上,进一步发展出了ZINB。
4 Zero-inflated Negative Binomial
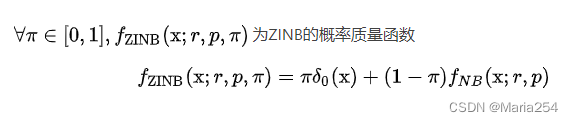

5 Zero-inflated (UMI based or read based).
已经有很多工作证明了对于UMI based sequencing来说, NB其实可以很好的刻画scRNA-seq data(可以参考Nancy Zhang的SAVER)。所以到底用ZINB还是NB还是要取决于测序的技术。不过目前大部分测序都是UMI-based了,所以NB可能会成为更general的选择
这篇关于单细胞数据分布 ZINB的理解的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!







