本文主要是介绍R与bioconductor--ExpressionSet SummarizedExperiment GEOquery biomaRt S4-Classes S4-Methods,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
博主自学了coursera上来自约翰霍普金斯大学<使用Bioconductor分析基因组科学数据>,很不错,推荐给大家
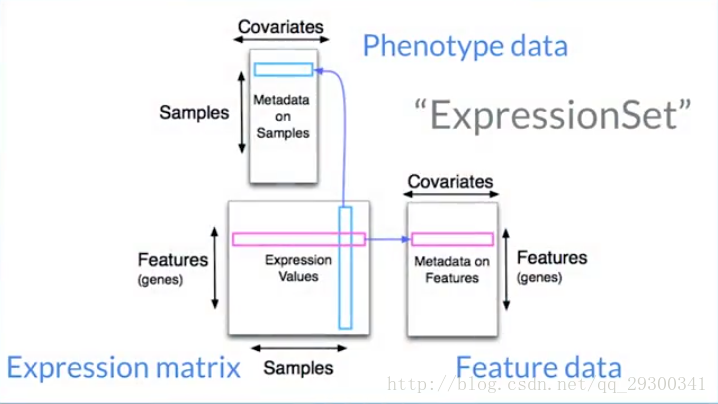
ExpressionSet Overview
ExpressionSet
-Expression Matrix
-Phenotype data Feature data
eSet:仅仅是有多个Expression Matrix
ExpressionSet
library(ALL)
data(ALL)
experimentData(ALL)
exprs(ALL)[1:4,1:4]
head(sampleNames(ALL))
head(featureNames(ALL))
head(pData(ALL))
ALL$sex
ALL[1:10,1:5]
featureData(ALL)#包含关于基因的信息,但是经常没有
ids <- featureNames(ALL)[1:5]
library(hgu95av2.db)#之前数据集里面写了芯片平台hgu95av2.db
as.list(hgu95av2ENTREZID[ids])
phenoData(ALL)#不如使用pData(ALL),phenoData(ALL)内容更多些
names(pData(ALL))#有时相当于varLabels(ALL),有时varLabels(ALL)更详细
SummarizedExperiment
#SummarizedExperiment
library(airway)
data("airway")
airway
colData(airway)#pData(ALL)
airway$cell
colnames(airway)
head(rownames(airway))
assayNames(airway)#要想获得表达矩阵,使用assay accessor,用assayNames()获得全部表达矩阵名
assay(airway,"counts")[1:4,1:4]
length(rowRanges(airway))
rowRanges(airway)#SummarizedExperiment特别之处在于每行每列都有关联的GRanges
start(airway)
gr = GRanges("1",ranges = IRanges(start = 1,end = 10^7))
subsetByOverlaps(airway,gr)
#'正是有着关联GRanges,可以取出染色体"某一区域内"基因的表达值
GEOquery
library(GEOquery)
eList <- getGEO("GSE11675")
length(eList)
eData = eList[[1]]
eData
names(pData(eData))
eList2 = getGEOSuppFiles("GSE11675")#下载原始tar包
eList2
biomaRt
library(biomaRt)
head(listMarts())
mart <- useMart("ensembl")
mart
head(listDatasets(mart))
ensemble <- useDataset("hsapiens_gene_ensembl",mart)
values <- c("202763_at","209310_s_at","207500_at")
getBM(attributes = c("ensembl_gene_id","affy_hg_u133_plus_2"),
filters = "affy_hg_u133_plus_2",values = values,mart = ensemble)
attributes <- listAttributes(ensemble)#可以查询到的条目
nrow(attributes)#可以把一个物种的基因转换到另一个物种的同源基因
head(attributes)
filters <- listFilters(ensemble)#可以查询到的条目
nrow(filters)#可以把一个物种的基因转换到另一个物种的同源基因
head(filters)
attributePages(ensemble)#attributes存储在一个一个page中,可以用这个减小搜索范围
attributes <- listAttributes(ensemble,page = "feature_page")
nrow(attributes)
R S4 Classes
library(ALL)
library(GenomicRanges)
#'S3对象就是像一个list,list中每个对象都有各自的name
#'而S4对象定义了每个class应该是有些什么东西
data("ALL")
ALL
class(ALL)
isS4(ALL)
class?ExpressionSet#查看一个class的简介
?"ExpressionSet-class"#查看一个class的简介
#'list的规则:首字母大写
#'构造方法
ExpressionSet()
getClass("ExpressionSet")#Slots插槽,就是这个class由哪儿些小class构成
ALL@annotation
annotation(ALL)
#class升级了,定义改变了,用updateObject
OLD_OBJECT = updateObject(OLD_OBJECT)
validObject(ALL)#检测对象是否正确,是否符合class的定义
R S4 Methods
library(GenomicRanges)
GenomicRanges::as.data.frame#S4方法
base::as.data.frame#S3方法
showMethods("as.data.frame")#可以看见,X类型不同,后续选用的程序代码也不同
#查看传入某一特定类型,对应的相关程序代码
getMethod("as.data.frame","GenomicRanges")
getMethod("as.data.frame",signature(x="GenomicRanges"))
#查看传入某一特定类型,对应的帮助文档
method?"as.data.frame,DataFrame"
method?"as.data.frame,GenomicRanges"
?"as.data.frame,DataFrame-method"
?"as.data.frame,GenomicRanges-method"
showMethods("findOverlaps")
getMethod("findOverlaps",signature(query = "Ranges",subject = "Ranges"))
?"findOverlaps,Ranges,Ranges-method"
#'S4缺点:难以找到help文档,难以直接看源代码,难以debug
#'但是最好S4写一个package,方便管理
最后是完整代码片段
#ExpressionSet
library(ALL)
data(ALL)
experimentData(ALL)
exprs(ALL)[1:4,1:4]
head(sampleNames(ALL))
head(featureNames(ALL))
head(pData(ALL))
ALL$sex
ALL[1:10,1:5]
featureData(ALL)#包含关于基因的信息,但是经常没有
ids <- featureNames(ALL)[1:5]
library(hgu95av2.db)#之前数据集里面写了芯片平台hgu95av2.db
as.list(hgu95av2ENTREZID[ids])
phenoData(ALL)#不如使用pData(ALL),phenoData(ALL)内容更多些
names(pData(ALL))#有时相当于varLabels(ALL),有时varLabels(ALL)更详细#SummarizedExperiment
library(airway)
data("airway")
airway
colData(airway)#pData(ALL)
airway$cell
colnames(airway)
head(rownames(airway))
assayNames(airway)#要想获得表达矩阵,使用assay accessor,用assayNames()获得全部表达矩阵名
assay(airway,"counts")[1:4,1:4]
length(rowRanges(airway))
rowRanges(airway)#SummarizedExperiment特别之处在于每行每列都有关联的GRanges
start(airway)
gr = GRanges("1",ranges = IRanges(start = 1,end = 10^7))
subsetByOverlaps(airway,gr)
#'正是有着关联GRanges,可以取出染色体"某一区域内"基因的表达值library(GEOquery)
eList <- getGEO("GSE11675")
length(eList)
eData = eList[[1]]
eData
names(pData(eData))
eList2 = getGEOSuppFiles("GSE11675")#下载原始tar包
eList2library(biomaRt)
head(listMarts())
mart <- useMart("ensembl")
mart
head(listDatasets(mart))
ensemble <- useDataset("hsapiens_gene_ensembl",mart)
values <- c("202763_at","209310_s_at","207500_at")
getBM(attributes = c("ensembl_gene_id","affy_hg_u133_plus_2"),filters = "affy_hg_u133_plus_2",values = values,mart = ensemble)
attributes <- listAttributes(ensemble)#可以查询到的条目
nrow(attributes)#可以把一个物种的基因转换到另一个物种的同源基因
head(attributes)
filters <- listFilters(ensemble)#可以查询到的条目
nrow(filters)#可以把一个物种的基因转换到另一个物种的同源基因
head(filters)
attributePages(ensemble)#attributes存储在一个一个page中,可以用这个减小搜索范围
attributes <- listAttributes(ensemble,page = "feature_page")
nrow(attributes)library(ALL)
library(GenomicRanges)
#'S3对象就是像一个list,list中每个对象都有各自的name
#'而S4对象定义了每个class应该是有些什么东西
data("ALL")
ALL
class(ALL)
isS4(ALL)
class?ExpressionSet#查看一个class的简介
?"ExpressionSet-class"#查看一个class的简介
#'list的规则:首字母大写
#'构造方法
ExpressionSet()
getClass("ExpressionSet")#Slots插槽,就是这个class由哪儿些小class构成
ALL@annotation
annotation(ALL)
#class升级了,定义改变了,用updateObject
OLD_OBJECT = updateObject(OLD_OBJECT)
validObject(ALL)#检测对象是否正确,是否符合class的定义library(GenomicRanges)
GenomicRanges::as.data.frame#S4方法
base::as.data.frame#S3方法
showMethods("as.data.frame")#可以看见,X类型不同,后续选用的程序代码也不同
#查看传入某一特定类型,对应的相关程序代码
getMethod("as.data.frame","GenomicRanges")
getMethod("as.data.frame",signature(x="GenomicRanges"))
#查看传入某一特定类型,对应的帮助文档
method?"as.data.frame,DataFrame"
method?"as.data.frame,GenomicRanges"
?"as.data.frame,DataFrame-method"
?"as.data.frame,GenomicRanges-method"showMethods("findOverlaps")
getMethod("findOverlaps",signature(query = "Ranges",subject = "Ranges"))
?"findOverlaps,Ranges,Ranges-method"
#'S4缺点:难以找到help文档,难以直接看源代码,难以debug
#'但是最好S4写一个package,方便管理这篇关于R与bioconductor--ExpressionSet SummarizedExperiment GEOquery biomaRt S4-Classes S4-Methods的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!





