
尝试着将神经网络的元件与生物学意义联系起来。大胆假设,小心求证!
PMC | Genome Res. | GitHub
下载
从ENCODE Project Consortium下载125种细胞类型的数据。
从Roadmap Epigenomics Consortium下载39种细胞类型的数据。
数据形式为DNase-seq的peak信息,保存在BED格式的文件中。
使用未去重叠(overlap)的peak数据。
预处理
- 以1%的FDR使用模拟方法修改原始数据集——robustness
- 归并重叠的peaks共 $2,071,886$ 个峰,比对到hg19参考基因组
- 标准输入为每个位点600bp的DNA序列长度
- 标准标签为一个164维的二值向量,该向量表示这个峰(位点)在164种细胞类型中的开放情况(1为开放,0为不开放)
- 将数据集切分为训练集、测试集和验证集
- training data:训练模型参数
- testing data:计算序列特异性参数
- validation data:用于early stopping
- 应模型后续分析需要,使用GENOME v18 reference catalog将位点分为promoter(转录起始位点周围2kb区域)、intragenic(与基因区域发生重叠)、intergenic(位于基因间区域内)三类
模型
训练模型和测试模型的用途有细微差别。
训练
训练模型是一个3CNN+2FC的简单神经网络,此项目的亮点不在神经网络的设计,而在于将神经网络的元件与生物学意义结合起来,这也是以往项目中我所想不透的地方。训练网络如下: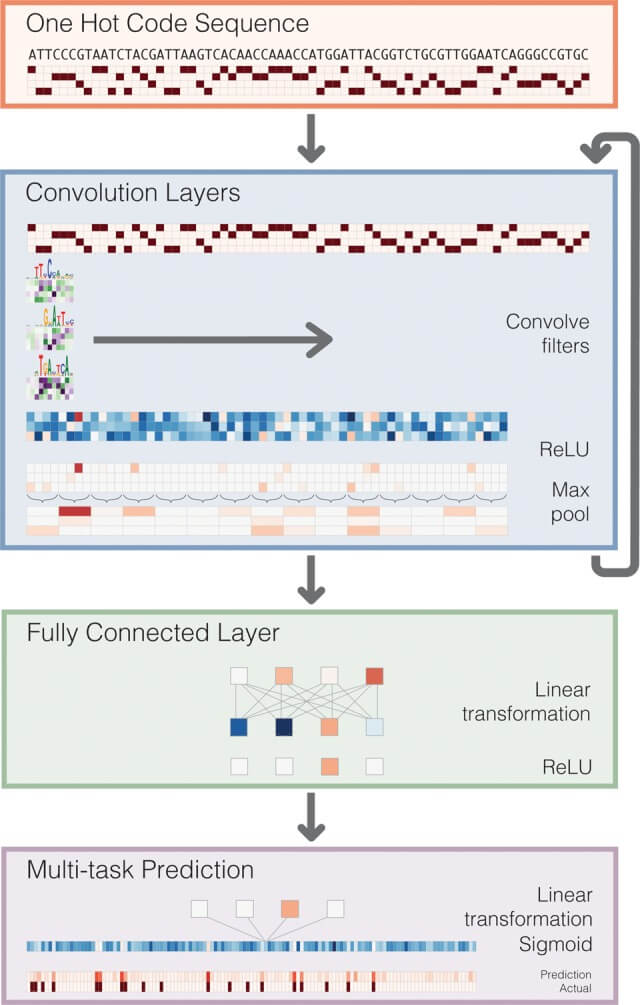
模型的输入是一个one-hot编码的 $4 times 600$ 的序列(与处理中已经将所有位点的序列长度截取到了600bp)。
the first CNN是本模型的重点,它包括了一个卷积层、一个激活层和一个池化层。
卷积层使用的是300个 $4 times M$ 的一维卷积核(filter,滤波器),其中 $M$ 长度跟motif长度相当(这里取19b)。作者在这里给每个filter赋予了生命力,认为它们不仅是一种网络元件。因为每个卷积核是基于所有序列优化得来的,所以我们认为卷积核代表了所有序列共有的一种信息,即模式。这是符合思考逻辑的。
简而言之,每个卷积核可能代表了一种motif,这个motif具体的生物学意义未知。此项目试图从蛋白质结合motif出发去验证卷积核中是否存在相应的结构与之对应。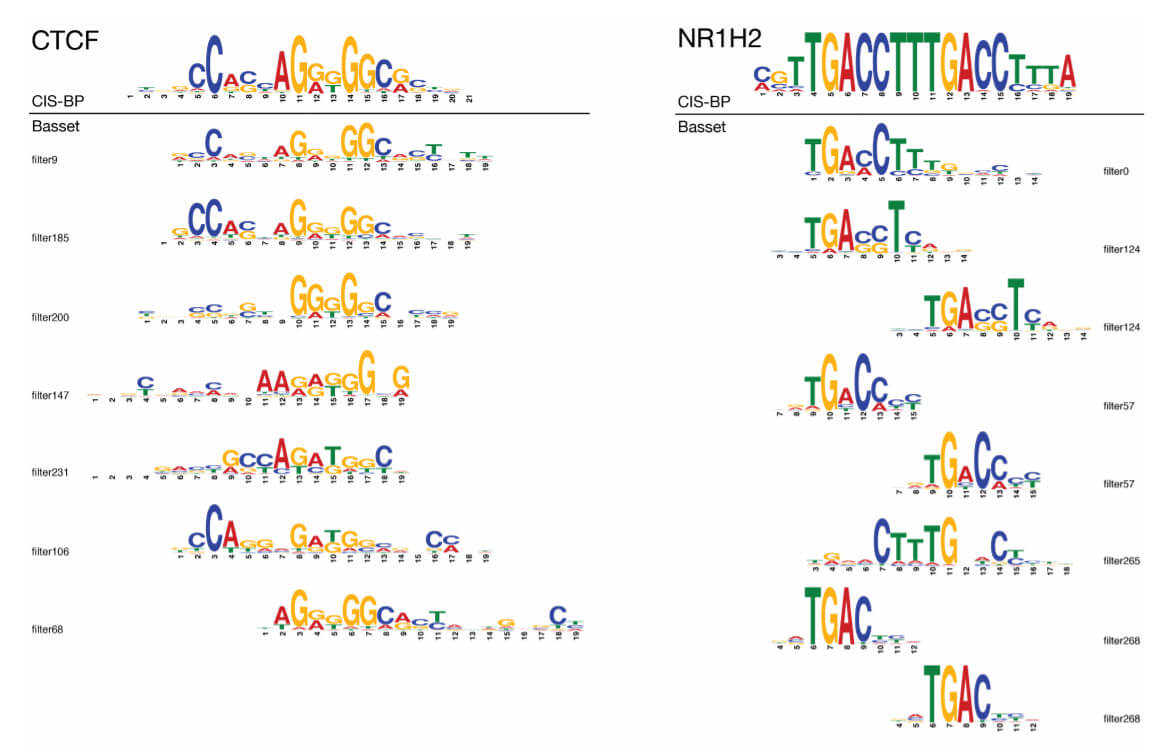
这是一种不错的假设-验证思想。
模型的最后一层也就是第二个全连接层的输出是164个节点,分别代表了该600bp的序列在164种类型细胞种的开放情况。参考标准标签计算binary loss进行参数优化。
测试
测试阶段作者做了很有意思的事情。
为了计算卷积核(motif)对(预测的)开放性打分结果的影响程度,将第一CNN的输出结果(每个卷积核会将 $4 times 600$ 的序列转化为(600-19+1)维的向量)全部用其平均值替换,这将消除卷积核的特异性影响。用替换结果进行后续计算,将计算结果与未经替换的预测结果进行比较。计算差异的平方和,这个值作为该卷积核的influence值。
IC(Information Content)值得原文计算方法如下:
$$
mathrm{IC} = sum_{i,j}{m_{ij}log_2 m_{ij}} - sum_{i,j}{ b_j log_{2}b_{j}}
$$
写成这样似乎更清楚点:
$$
mathrm{IC} = sum_{i=1}^{19} sum_{j=1}^4 m_i^{(j)} log_2 m_i^{(j)} - 19sum_{j=1}^4 b^{(j)} log_2 b^{(j)}
$$
其中向量 $b$ 是四个碱基分布的背景值,向量 $m;(4 times 19)$ 是motif各个位置的碱基概率分布,等效于一个卷积核。
有趣的是,除了对卷积核进行上述操作计算IC值influence值,作者还直接从数据库中(CIS-BP数据库)下载已知的蛋白质motif代替卷积核进行计算,最后一同进行比较分析。





