本文主要是介绍基因集合打分不考虑基因高低表达 基因集合评分不考虑高低表达 绝对值 基因集合里面的正负表达影响addmodulescore的结果 构建的基因集进行评分时 基因的高低正负表达基因集合评分不考虑高低表达,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
GSEA分析一文就够(单机版+R语言版)
8种方法可视化你的单细胞基因集打分 - 腾讯云开发者社区-腾讯云 (tencent.com)
GSEA的统计学原理试讲
一、开发背景
该算法最初开发是受microarray RNA数据驱动,旨在解释基因组数据,获得相较于单个基因更加深入的生物学见解。
基于差异基因分析存在的缺陷:
- 多重假设检验校正后,只有少量基因甚至没有基因的差异达到统计学显著性。【生物学差异被技术噪音,如microarray,所掩盖】
- 得到大量的统计上显著的差异表达基因,但由于缺乏或未按统一生物学主题对基因进行组织,对结果的解释相当困难,依赖于生物学家强大的生物学背景。
- 聚焦于单基因差异,会忽略一些真正有生物学意义的生物通路。如,(基因协同发挥作用)编码代谢途径的成员的所有基因增加20%可能会显著改变细胞的代谢过程,这可能比单个基因增加20倍更重要。
- 同一生物学系统,不同研究团队获得差异基因列表可能差别巨大,甚至很少重叠。
GSEA vs. DEGs
- DEGs多聚焦于单个基因;GSEA是在基因集水平上比较不同生物学系统(不同样本)间的转录组差异。
- 结果稳健性更好,在不同团队研究结果中的生物学意义的可重复性和解释性更好。
- 高度灵活性,主要体现在基因集的来源。可以使用公共数据库如MsigDb,还可以根据研究目的自己构建。
二、数学原理:
总的来说,GSEA富集分析有以下三个要点:
- input data1 :样本的全基因组RNA测序数据及样本的表型标签信息【样本数尽量多一些,否则假阳性高】
- input data 2:基于某个合适的指标(如差异倍数FC)对所有基因排序,获得排序基因列表
L = {g1, g2, g3, g4, …… gN};【可根据研究需要,制定个性化排序方案,如基于与兴趣TF的相关性。】 - input data 3:基因集S【具有充分个性化空间。可自己定义,也可以人工筛选和矫正。】
- input data 4:指定计算过程中权重值p。
ES的数学计算过程如下:
总的原则:看某个基因集S的基因在L上随机分布 or 分布在顶部 or 分布在尾部。【为GSEA算法的核心,数学原理见下】
- 计算富集分数(Enrichment Score, ES)
- 解读:绝对值越大,代表基因集S在排名列表L的顶部(正值)或底部(负值)被过度代表(overrepresented);
- 计算过程:给定
初始统计量x, 沿着排序列表从top→bottom遍历每个基因,遇到属于基因集S的基因,则x = x+ai;若遇到不属于基因S的基因j,则x = x - aj…… 直到遍历所有基因。【注意:a的值取决于基因i或基因j与分组性状的关联程度,关联越大,则a越大;反之,则越小】。ES即为该过程中的x的最大取值,对应加权的Kolmogorov-Smirnov样的统计量(ref7)。
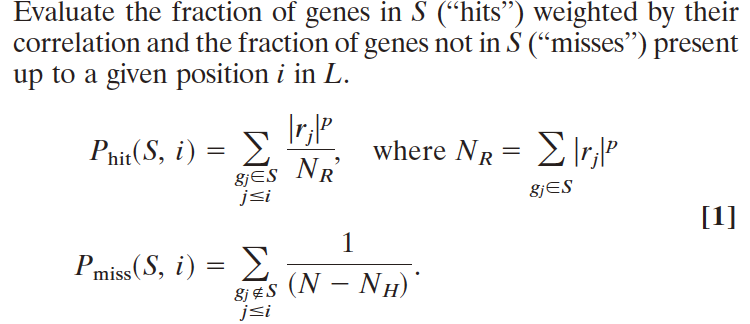
图例:①Phit= 基因集S中的加权和,Pmiss = 非基因集S中的加权和。②ES = Phit - Pmiss。②
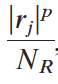
对应ai部分。p代表加权,若p=0,则公式简化为标准的Kolmogorov-Smirnov统计量;若p=1,则Phit的分母为基因集S中所有基因与性状的关联强度之和,基因集S中每个基因与该性状的关联都对该和进行标准化处理。【r代表基因与兴趣性状之间的关联强度,可以由FC等来评估】
- ES的统计显著性评估
- 统计学检验方法:基于经验表型的置换检验方法(empirical phenotype-based permutation test);
- 构建零分布:对每个样本重新分配表型标签、重新排序所有基因、重新计算基因集S的ES值;以上过程重复1000次,该1000个ES值构成零分布(null distribution);
- 计算P值:绘制直方图,p值即为ESnull distribution中≥或≤ESobserved的比例。该p值为经验名义p值。
- 结果解读:小于α值(如0.05),则拒绝零假设,认为基因集S在排序列表L的top端或bottom端富集;若≥α值,则接受零假设,认为兴趣基因集S内基因在排序列表L中随机分布。
- p值的多重假设检验校正
- 适用场景:当对多个基因集S(S1,S2, S3, S4……)进行GSEA分析时;
- 计算过程:①基于基因集S大小,对ES进行标准化处理,获得NES值;②对每个NES计算FDR值。
- 结果解读:FDR值代表,给定NES值对应的基因集为假阳性(富集)的估计概率;值越大,则假阳性的概率越大。
三、生物学意义的挖掘
- 富集通路的分析(略)。
S1:基因集S1主要分布在排序列表的top端,ES分值较高,p值显著;
S2:基因集S2在排序列表中随机分布,ES值低,p值不显著;
S3:基因集S3非随机分布,但也并不在top or bottom呈现集中分布模式,ES值较高,但p值不显著。
因此,富集分析的结果结果,要综合考虑p值及ES值两个指标。

- leading-edge subset
- 背景:可以通过多种方法定义基因集;需要注意的是,并不是基因集内的每个基因成员都会参与到兴趣的生物学通路中。
- 定义:基因集S中位于x最大值(偏离0值最大的位置)之前的基因(包含最大值位置对应的基因)。
- 结果解读:①代表基因集S中对ES值贡献最大的一小簇基因(核心基因);②代表基因集中在兴趣生物学通路中发挥关键作用的核心成员。
- 应用示例:
- 如下图,作者通过对p53突变和p53野生型的转录组数据进行GSEA富集分析,发现to3富集的信号通路(按p排序)为Ras信号通路、Ngf信号通路、Igf1信号哦通路。进而分析对三个通路富集贡献最大的基因,发现有四个MAPK信号通路相关的基因对该三个通路的富集均产生较大贡献。从而可以预测,MAPK信号通路中存在亚通路在p53-肿瘤组织中发挥重要作用。

四、实现方式
R包:clusterProfiler,需要自己做完差异分析,得到deg这个数据库,它有一列是logFC,有一列是基因的名字(这里举例是symbols),然后就可以无缝运行下面的代码啦!
我的示例的deg来源于单细胞分析的FindMarkers函数,代码如下;
Idents(sce)='singleR'
deg=FindMarkers(object = sce, ident.1 = 'Fibroblasts',ident.2 = 'Fibroblasts activated',min.pct = 0.01, logfc.threshold = 0.01,thresh.use = 0.99)
head(deg)复制
在msigdb数据库网页可以下载全部的基因集,我这里方便起见,仅仅是下载 h.all.v7.2.symbols.gmt文件:
### 对 MsigDB中的全部基因集 做GSEA分析。
# http://www.bio-info-trainee.com/2105.html
# http://www.bio-info-trainee.com/2102.html
# https://www.gsea-msigdb.org/gsea/msigdb
# https://www.gsea-msigdb.org/gsea/msigdb/collections.jsp
{ geneList= deg$avg_logFC names(geneList)= toupper(rownames(deg))geneList=sort(geneList,decreasing = T)head(geneList)library(ggplot2)library(clusterProfiler)library(org.Hs.eg.db)#选择gmt文件(MigDB中的全部基因集)gmtfile ='MSigDB/symbols/h.all.v7.2.symbols.gmt'# 31120 个基因集#GSEA分析library(GSEABase) # BiocManager::install('GSEABase')geneset <- read.gmt( gmtfile ) length(unique(geneset$term))egmt <- GSEA(geneList, TERM2GENE=geneset, minGSSize = 1,pvalueCutoff = 0.99,verbose=FALSE)head(egmt)egmt@result gsea_results_df <- egmt@result rownames(gsea_results_df)write.csv(gsea_results_df,file = 'gsea_results_df.csv')library(enrichplot)gseaplot2(egmt,geneSetID = 'HALLMARK_EPITHELIAL_MESENCHYMAL_TRANSITION',pvalue_table=T)gseaplot2(egmt,geneSetID = 'HALLMARK_MTORC1_SIGNALING',pvalue_table=T)
}复制
参考文献:
Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles
Aravind Subramanian, Pablo Tamayo, Vamsi K. Mootha, Sayan Mukherjee, Benjamin L. Ebert, Michael A. Gillette, Amanda Paulovich, Scott L. Pomeroy, Todd R. Golub, Eric S. Lander, Jill P. Mesirov Proceedings of the National Academy of Sciences Oct 2005, 102 (43) 15545-15550; DOI: 10.1073/pnas.0506580102
这篇关于基因集合打分不考虑基因高低表达 基因集合评分不考虑高低表达 绝对值 基因集合里面的正负表达影响addmodulescore的结果 构建的基因集进行评分时 基因的高低正负表达基因集合评分不考虑高低表达的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!



