本文主要是介绍RNAseq分析:Step6(计算表达丰度),希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
目录
前记
一、计算FPKM
二、计算reads数
后记
前记
RNA-seq技术是研究基因表达的常用方法之一,其表达丰度计算是RNA-seq数据分析的重要步骤之一。
RNA-seq表达丰度计算的基本流程如下:
-
序列比对:将测序数据比对到参考基因组,得到每个基因的计数。
-
转录本重构:使用转录本拼接软件,如Cufflinks或StringTie,将比对后的 Bam/Sam 文件转换为每个转录本的表达值。这里的转录本可能是已知的基因、未知的基因或转录本。
-
表达值的归一化:考虑样本间的技术差异和表达量大小的影响,对表达值进行归一化。常用的归一化方法包括RPKM、FPKM、TPM等,其中TPM是近年来推出的一种比较推荐的归一化方法。
-
差异表达分析:通过比较不同样本下的基因或转录本表达值,识别差异表达的基因或转录本。差异表达分析常用的软件包括DESeq2、edgeR和limma等。
-
基因本体注释和通路分析:将差异表达的基因或转录本进行功能注释,通常使用基因本体注释(GO)和通路分析(KEGG)等方法。这一步有助于研究人员理解基因在生物学过程中的功能和调控机制。
总的来说,RNA-seq表达丰度计算需要经历序列比对、转录本重构、表达值的归一化、差异表达分析和功能注释等步骤。这些步骤需要使用不同的软件和工具,根据实验设计和分析目的合理选择并组合使用。
本文主要介绍如何使用stringtie软件计算FPKM值以及如何利用HTSeq-count软件计算reads数目。
一、计算FPKM
使用stringtie计算基因和转录本的表达丰度。
#使用stringtie计算基因和转录本的FPKM
stringtie -e -p 2 -G ~/rnaseq/tair10_genome/tair10.gtf -A SRR3418005_genes.gtf -o SRR3418005_transcripts.gtf ~/rnaseq/hisat2_results/SRR3418005.bam &
每个bam文件处理之后会得到两个gtf文件,分别是genes.gtf文件和transcripts.gtf文件,文件中包含,基因或转录本的FPKM值信息。
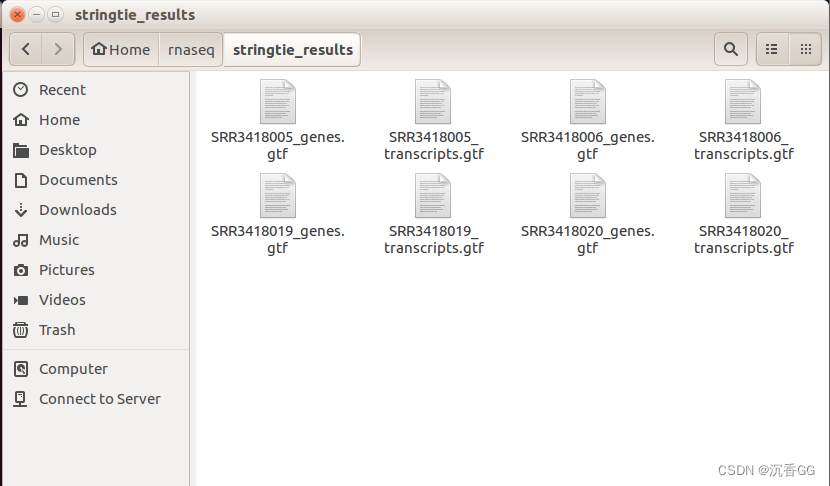
将得到四个样本的基因和转录本的gtf文件分别进行合并。
#删除第一行标题,以下步骤将四个gtf文件整合为一个
sed -i '1d' *_genes.gtf #排序并输出到merge文件夹下
mkdir merge
sort SRR3418005_genes.gtf > merge/SRR3418005_genes.gtf
sort SRR3418006_genes.gtf > merge/SRR3418006_genes.gtf
sort SRR3418019_genes.gtf > merge/SRR3418019_genes.gtf
sort SRR3418020_genes.gtf > merge/SRR3418020_genes.gtf#切换到merge文件夹,操作如下
cd merge
join -t $'\t' SRR3418005_genes.gtf SRR3418006_genes.gtf | join - SRR3418019_genes.gtf | join - SRR3418020_genes.gtf > out_fpkm.gtf
awk -F ' ' '{print $1"\t"$2"\t"$3"\t"$4"\t"$5"\t"$5"\t"$6"\t"$7"\t"$8"\t"$16"\t"$24"\t"$32}' out_fpkm.gtf > fpkm.gtf #fpkm_gtf为最终的FPKM注释文件
二、计算reads数
#计算SRR3418005样本基因的counts数目
htseq-count -q -f bam -s no -i gene_id ~/rnaseq/hisat2_results/SRR3418005.bam ~/rnaseq/tair10_genome/tair10.gtf > SRR3418005.count &计算过程如下所示:
整合四个样本的count文件。
#count文件整合
join SRR3418005.count SRR3418006.count | join - SRR3418019.count | join - SRR3418020.count > count.txt
sed -i 's/ /\t/g' count.txt后记
以上是FPKM值计算和reads数计算的方法,后续会利用count.txt文件进行差异表达分析。
2023.8.24
----CXGG
这篇关于RNAseq分析:Step6(计算表达丰度)的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!







