丰度专题
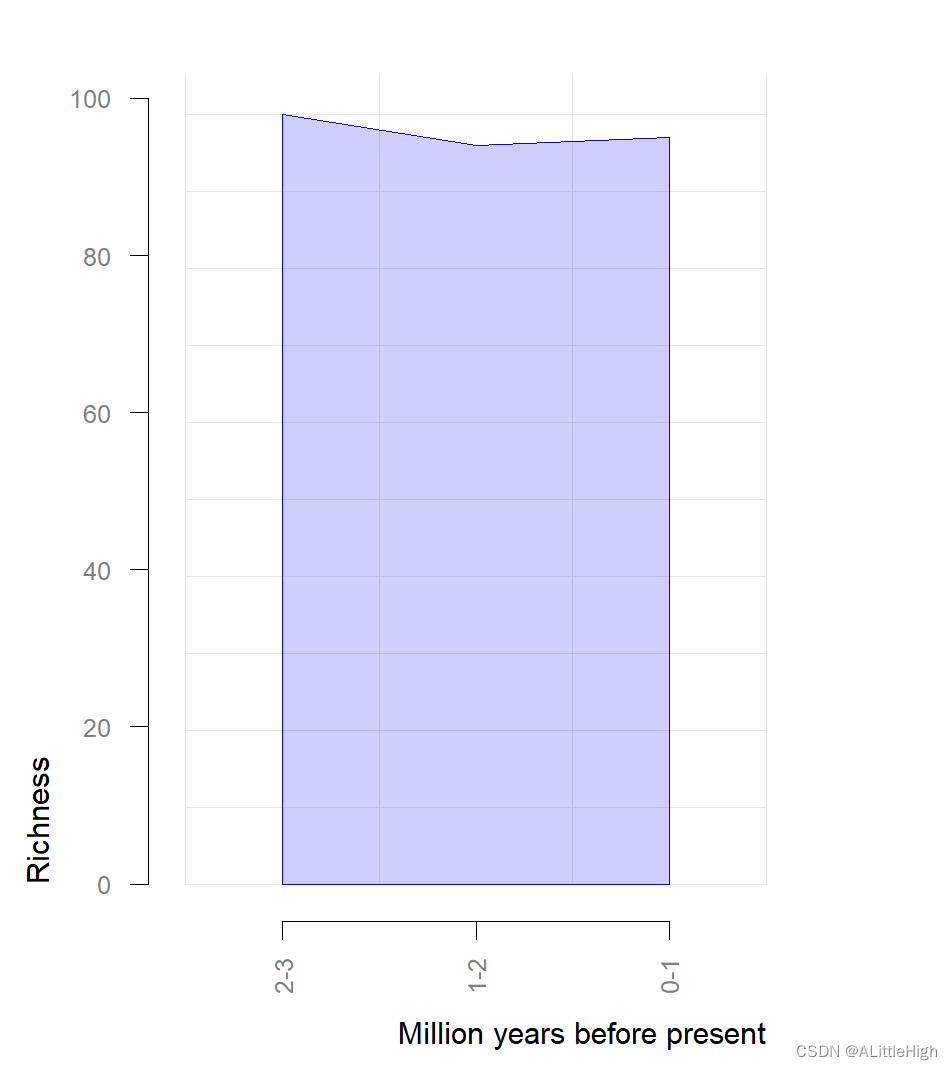
R语言【paleobioDB】——pbdb_richness():绘制指定类群的数量丰度
Package paleobioDB version 0.7.0 paleobioDB 包在2020年已经停止更新,该包依赖PBDB v1 API。 可以选择在Index of /src/contrib/Archive/paleobioDB (r-project.org)下载安装包后,执行本地安装。 Usage pbdb_richness (data, rank, res, te
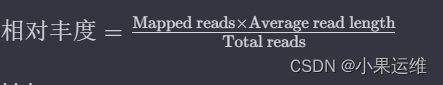
基于BWA,Bowtie2,samtools、checkm等工具计算宏基因组学序列分析中Contigs与Genes在样品中的丰度,多种计算方式和脚本对比
计算contigs和genes相对丰度可以提供有关微生物群落结构和功能的信息。以下是计算这两个指标的意义: 1. Contigs的相对丰度:contigs是利用基因组测序技术获得的碎片序列,通过计算contigs的相对丰度可以了解微生物群落中不同菌种的相对丰度。这可以帮助研究者理解微生物群落的物种组成和群落结构。 2. Genes的相对丰度:基因是生物体内功能的基本单位,通过计算基因的相对丰度
RNAseq分析:Step6(计算表达丰度)
目录 前记 一、计算FPKM 二、计算reads数 后记 前记 RNA-seq技术是研究基因表达的常用方法之一,其表达丰度计算是RNA-seq数据分析的重要步骤之一。 RNA-seq表达丰度计算的基本流程如下: 序列比对:将测序数据比对到参考基因组,得到每个基因的计数。 转录本重构:使用转录本拼接软件,如Cufflinks或StringTie,将比对后的 Bam/Sam 文