本文主要是介绍maftools癌症体细胞变异(突变)分析工具学习,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
Maftools 是一个专门用于分析和可视化突变数据的 R 包。全名为 "Mutation Annotation Format Tools",它主要处理 MAF(Mutation Annotation Format)文件,这种文件格式广泛用于存储和共享癌症基因组中的突变数据。 Maftools 的主要功能包括:
-
突变数据解析:可以方便地加载 MAF 文件,并将其解析为 R 中的对象,便于进一步分析。
-
突变数据可视化:提供了多种强大的可视化工具,如瀑布图(Waterfall plot)、突变频率图、共突变和互斥突变分析、突变频谱分析等,帮助研究人员更直观地理解突变数据的特征。
-
临床数据整合:可以将临床数据与突变数据整合,分析不同临床亚群的突变特征和生存分析等。
-
统计分析:包括突变负荷分析、通路富集分析、TCGA数据整合分析等功能。
详细的MAF数据格式解释在官网:https://docs.gdc.cancer.gov/Data/File_Formats/MAF_Format/

截图展示部分内容,如果需要了解基因组数据相关概念的可以登录该网站细细研究一下。
那么得到这部分数据我们可以做点什么呢?首先是根据突变情况联合各种临床参数;其次现已经进入了免疫治疗时代,可以将其与免疫治疗的数据联合分析;或者如果重点要研究某一个分子的突变情况,研究者可以先来看看这里的突变数据~ 总之,用途还是非常多的!
在使用之前建议先熟读一下官方文档,比笔者展示的更细致全面!链接:https://bioconductor.org/packages/release/bioc/vignettes/maftools/inst/doc/maftools.html#1_Introduction
分析流程
1、导入(使用TCGAmutations包下载突变数据)
清洗好的LIHC数据
rm(list = ls())
library(maftools)
# TCGAmutations包含了已经预处理好的TCGA的各个癌种的突变数据(不包括CNV)
library(TCGAmutations)
library(tidyverse)
load("TCGA-LIHC_sur_model.Rdata")
head(exprSet)[1:5,1:5]
# TCGA-FV-A495-01A TCGA-ED-A7PZ-01A TCGA-ED-A97K-01A TCGA-ED-A7PX-01A TCGA-BC-A3KF-01A
# WASH7P 1.913776 1.2986076 1.967382 1.586170 0.8210928
# AL627309.6 3.129116 0.5606928 3.831265 1.363539 1.2461127
# WASH9P 2.490476 2.8140204 2.960338 2.106464 1.5945195
# MTND1P23 2.773335 3.4114257 2.591028 3.353850 2.9667012
# MTND2P28 4.668896 8.0896003 9.426092 9.610934 10.0257594
head(meta)
# ID OS OS.time race age gender stage T N M
# TCGA-FV-A495-01A TCGA-FV-A495-01A 0 0.03333333 WHITE 51 FEMALE II 2 NA 0
# TCGA-ED-A7PZ-01A TCGA-ED-A7PZ-01A 0 0.20000000 ASIAN 61 MALE II 2 NA 0
# TCGA-ED-A97K-01A TCGA-ED-A97K-01A 0 0.20000000 ASIAN 54 MALE III 3 0 0
# TCGA-ED-A7PX-01A TCGA-ED-A7PX-01A 0 0.20000000 ASIAN 48 FEMALE II 2 NA 0
# TCGA-BC-A3KF-01A TCGA-BC-A3KF-01A 0 0.26666667 WHITE 66 FEMALE I 1 NA 0
# TCGA-DD-A4NR-01A TCGA-DD-A4NR-01A 1 0.30000000 WHITE 85 FEMALE I 1 0 0
dim(meta)
# [1] 365 10
maf = tcga_load("LIHC")
dim(maf@data)
# [1] 36445 23
length(unique(maf@data$Tumor_Sample_Barcode))
# [1] 365
2、数据预处理
# 提取了每个样本的名称(前者是1:16位的/后者是全称)
samp = data.frame(ID = str_sub(unique(maf@data$Tumor_Sample_Barcode),1,16),long = unique(maf@data$Tumor_Sample_Barcode))
# merge() 函数会自动根据指定的列(如 ID 列)将两个数据框的数据进行配对合并
samp= merge(meta,samp,by = "ID")# 临床信息
# 其实上面已经准备好了,再来一次吧~
maf@clinical.data = left_join(maf@clinical.data,samp,by = c("Tumor_Sample_Barcode"="long"))
这里的临床信息很丰富。

3、oncoplot图
# subsetMaf()是maftools包中的一个函数,用于从原始MAF 数据中提取特定的样本或突变类型的子集。
maf = subsetMaf(maf,tsb = samp$long)
# --Possible FLAGS among top ten genes:
# TTN
# MUC16
# OBSCN
# -Processing clinical data
# 意味着它们是突变频率较高的基因# 定义突变参数
cla1 = c('Frame_Shift_Del','Missense_Mutation', 'Nonsense_Mutation', 'Multi_Hit', 'Frame_Shift_Ins','In_Frame_Ins', 'Splice_Site', 'In_Frame_Del')
# 上面的信息藏在variant.classification.summary中
cla2 = colnames(maf@variant.classification.summary)[-c(1,ncol(maf@variant.classification.summary))]
# 使用intersect函数找到cla1和cla2的交集,即共同的突变分类;然后使用setdiff函数找到cla2中不在cla1中的突变分类,将它们合并以形成最终的突变分类列表cla
cla = c(intersect(cla1,cla2),setdiff(cla2,cla1))
# 突变分类分配颜色
col = RColorBrewer::brewer.pal(n = length(cla), name = 'Paired')
names(col) = cla
# 定义调色板并分配给临床特征
fabcol = RColorBrewer::brewer.pal(n = 9,name = 'Spectral')racecolor = fabcol[1:3];names(racecolor) = na.omit(unique(maf@clinical.data$race))
stagecolor = fabcol[4:5];names(stagecolor) = na.omit(unique(maf@clinical.data$stage.x))
agecolor = fabcol[6:7];names(agecolor) = na.omit(unique(maf@clinical.data$age))
gendercolor = fabcol[8:9];names(gendercolor) = na.omit(unique(maf@clinical.data$gender))fabcolors = list(race = racecolor,stage = stagecolor,age = agecolor,gender = gendercolor)
oncoplot(maf = maf, colors = col, top = 20,clinicalFeatures = c("race","stage","age","gender"), sortByAnnotation = TRUE, annotationColor = fabcolors)# Following columns are missing from annotation slot of MAF. Ignoring them..
# [1] "stage" "age"
相关概念:
-
Frame Shift Del (框移缺失):在DNA序列中删除一个或多个碱基,而这种删除的数量不是三的倍数,导致从突变点起编码框架发生移位。这种帧移通常会导致完全不同的氨基酸序列和一个提前的终止密码子,从而产生功能丧失的蛋白质。
-
Missense Mutation (错义突变):在DNA序列中的一个点发生单个碱基的变化,导致编码一个不同的氨基酸。这种突变可以改变蛋白质的功能,有时导致疾病。
-
Nonsense Mutation (无义突变):在DNA序列的一个点发生变化,导致一个氨基酸的密码子变成了终止密码子。这导致蛋白质的提前终止,通常会产生一个非功能的或缺失关键功能域的蛋白质。
-
Multi Hit:指一个基因在同一细胞中发生多次突变。这通常指多个独立的突变事件影响同一个基因,可能导致该基因功能的完全丧失或显著改变。
-
Frame Shift Ins (框移插入):在DNA序列中插入一个或多个碱基,而这种插入的数量不是三的倍数,导致编码框架发生移位。这也会导致蛋白质序列的改变和可能的早终止。
-
In Frame Ins (同框插入):在DNA序列中插入的碱基数量是三的倍数,不会改变读码框,但会在蛋白质中添加额外的氨基酸。这种突变可能改变蛋白质的功能或稳定性,但蛋白质的其余部分仍然按原设计翻译。
-
Splice Site (剪接位点突变):这类突变发生在基因内编码剪接位点的区域,可能影响mRNA的剪接过程,导致畸形或功能失常的蛋白质。
-
In Frame Del (同框缺失):在DNA序列中删除的碱基数量是三的倍数,不改变读码框,但会在蛋白质中删除一些氨基酸。与同框插入类似,这种突变可能影响蛋白质的功能或稳定性,但不会导致读码框移位。

4、综合展示图
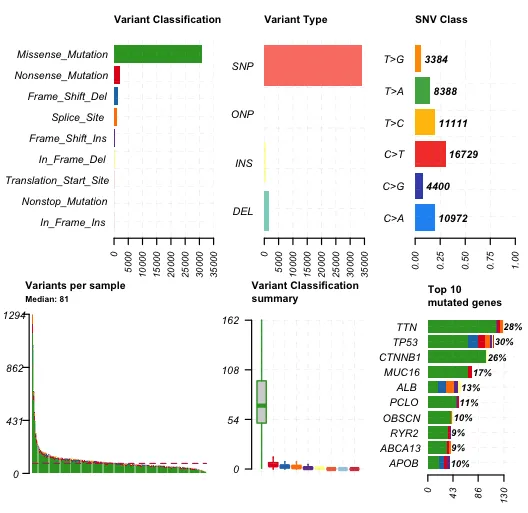
plotmafSummary(maf = maf,rmOutlier = TRUE,addStat = "median",dashboard = TRUE,titvRaw = FALSE)
相关概念:
-
SNP (Single Nucleotide Polymorphism,单核苷酸多态性):SNP是指在DNA序列中一个单一的碱基(核苷酸)在人群中存在两种或多种变体的情况。例如,一个位置的碱基可能在大多数人中是"A",但在某些人中可能是"G"。这种类型的遗传变异非常普遍,并且与许多遗传性疾病和个体之间的表型差异有关。
-
ONP (Oligonucleotide Polymorphism,少核苷酸多态性):ONP通常指的是在DNA序列中长度为2到几个核苷酸的短序列变异。这些可以视为较长的SNP或短小的插入和删除。ONP比单一的SNP更复杂,可以影响更多的生物学过程,例如蛋白质的编码或基因表达的调控。
-
INS (Insertion,插入):插入是指一个或多个核苷酸添加到DNA序列中的事件。插入的长度可以从一个碱基到几千个碱基不等。根据插入的大小和位置,它们可以对基因的功能产生显著影响,例如改变蛋白质的结构或调控基因的表达。
-
DEL (Deletion,删除):删除是指DNA序列中一个或多个核苷酸的丢失。与插入类似,删除也可以广泛变化其长度和发生的位置。删除可能导致基因失功能、编码蛋白质结构的改变或影响基因的调控。
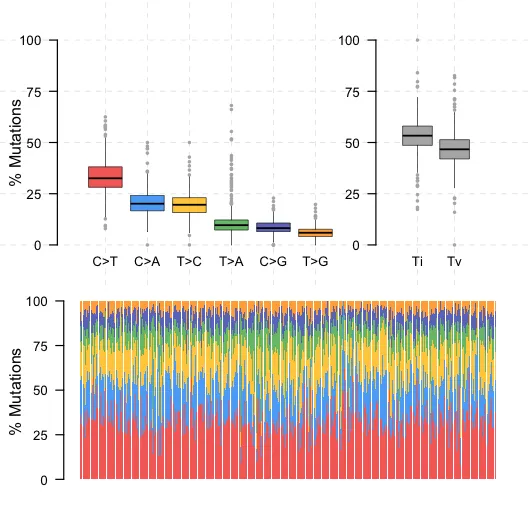
5、Ti/Tv比率
Ti/Tv比率(Transition/Transversion ratio)是指DNA序列中的碱基替换类型中,嘌呤(A/G)或嘧啶(C/T)之间的替换(称为转换,transitions)与嘌呤和嘧啶之间的替换(称为颠换,transversions)的比率。这个比率可以用来评估序列数据的质量,因为在人类基因组中,转换的发生率通常高于颠换。
● Transitions (Ti): 嘌呤之间的替换(A-G)或嘧啶之间的替换(C-T)
● Transversions (Tv): 嘌呤和嘧啶之间的替换(A-C, A-T, G-C, G-T)
titv <- titv(maf = maf,plot = F,useSyn = TRUE)
plotTiTv(res = titv)
6、雨景图
rainfallPlot(maf = maf, tsb = NULL, # 修改这里可以看特定样本 detectChangePoints = TRUE,pointSize = 0.4)
图中绘制了不同类型的点突变(单核苷酸多态性,SNP)在所有染色体上的分布,以及每种突变之间的距离(对数尺度)。
分布特点:通过观察不同染色体上的点分布,我们可以推断某些染色体可能存在更频繁的突变事件或特定类型的突变更为常见。
突变热点:某些区域可能显示出更密集的点,表明这些区域可能是突变热点,这些热点可能与特定的生物学性质或疾病相关。
突变类型的比较:不同颜色的分布可以帮助识别哪种类型的突变在该样本中更为常见。

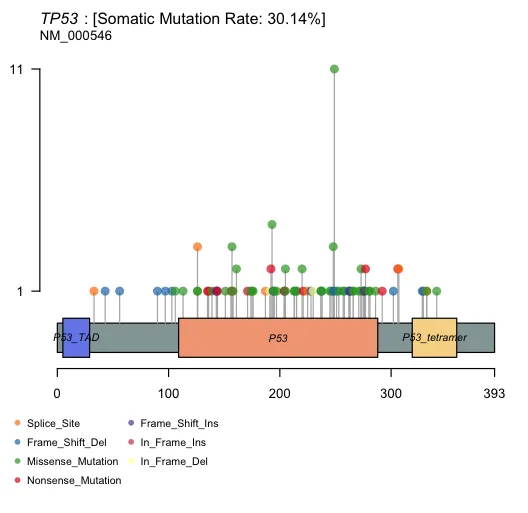
7、突变位点可视化
lollipopPlot(maf = maf,gene = "TP53",AACol="HGVSp_Short",showMutationRate = TRUE)
橙色区域表示TP53基因中的功能域(例如,P53和P53四聚体域),这些是蛋白质活性和功能的关键区域。
蓝色区域表示其他结构域,例如DBD_TAD域,与蛋白质的转录活性调节相关。
黄色区域表示TP53基因中的P53四聚体域(P53 tetramer)
突变集中区域:注意哪些区域有较多突变聚集,特别是功能域内的突变,因为这些可能影响蛋白质的功能。
突变类型:错义突变和无义突变的位置可以提供关于可能影响蛋白质功能的重要信息。例如,无义突变可能导致蛋白质提前终止。
频率高的突变:在图中垂直位置较高的突变表明在研究或数据集中观察到的频率较高,可能是该基因的关键突变。
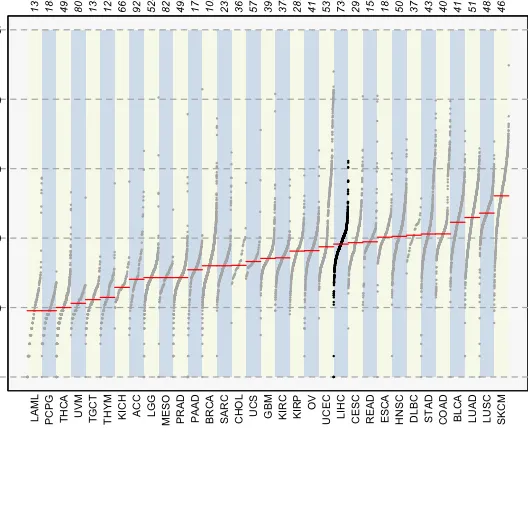
8、TCGA肿瘤突变负荷全景图
tcgaCompare(maf=maf.merge,cohortName="BREAST")
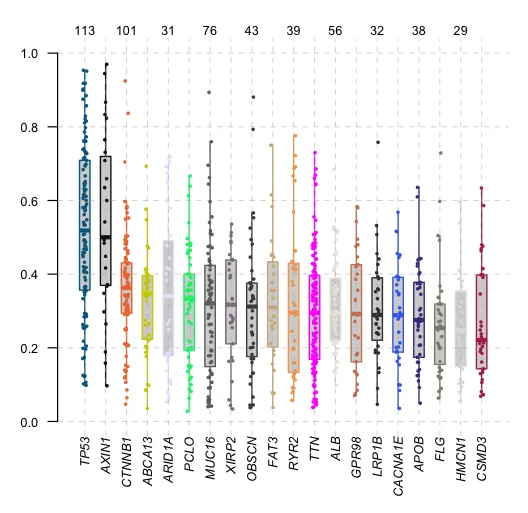
9、变异等位基因频率
plotVaf(maf = maf,top = 20)
# t_vaf field is missing, but found t_ref_count & t_alt_count columns. Estimating vaf..
t_vaf field is missing, but found t_ref_count & t_alt_count columns. Estimating vaf..:
这条信息说明原始MAF数据中缺少直接表示变异等位基因频率(Variant Allele Frequency,VAF)的字段(通常标记为 t_vaf),但是找到了代表参考等位基因计数(t_ref_count)和变异等位基因计数(t_alt_count)的列。VAF的计算公式通常是变异等位基因计数除以总等位基因计数(变异等位基因计数加上参考等位基因计数)
10、共现和互斥分析
somaticInteractions(maf,top = 20,genes = NULL,pvalue = c(0.05, 0.01),returnAll = TRUE,geneOrder = NULL,fontSize = 0.6,showSigSymbols = TRUE,showCounts = FALSE,countStats = "all",countType = "all",countsFontSize = 0.6,countsFontColor = "red",colPal = "BrBG",showSum = TRUE,plotPadj = FALSE,colNC = 9,nShiftSymbols = 5,sigSymbolsSize = 2,sigSymbolsFontSize = 0.8,pvSymbols = c(46, 42),limitColorBreaks = TRUE
)
基因标签:
● 横轴和纵轴都标有基因名,基因名旁的数字表示该基因突变的样本数量。
颜色和图例:
● 颜色深浅表示了P值的对数缩放(-log10 P-value),颜色越深(趋向绿色)表示统计上的关联性越强,颜色越浅(趋向黄色)表示关联性越弱。
● 绿色代表基因间的共现(co-occurrence),即这两个基因倾向于在同一样本中同时发生突变。
● 黄色代表基因间的互斥(mutually exclusive),即这两个基因倾向于不在同一样本中同时发生突变。
查看基因对的关系:图中每个单元格代表一对基因间的关系。例如,如果一个单元格位于基因A的行和基因B的列,那么这个单元格的颜色和标记描述了A和B之间的关系。
评估共现和互斥:找出那些颜色最深的绿色单元格,这些代表具有强烈共现关系的基因对;寻找黄色单元格,尤其是带有星号的,这些代表具有显著互斥关系的基因对。
统计显著性:特别关注那些带有星号的单元格,因为这些基因对显示了显著的共现或互斥关系。

11、基因突变状态与生存的关系
maf@clinical.data$OS.time <- as.numeric(maf@clinical.data$OS.time)
mafSurvival(maf = maf, genes = c('MUC16'), time = 'OS.time', Status = 'OS', isTCGA = TRUE)
# Looking for clinical data in annoatation slot of MAF..
# Number of mutated samples for given genes:
# MUC16
# 61
# Removed 13 samples with NA's
# Median survival..
# Group medianTime N
# <char> <num> <int>
# 1: WT 19.73333 352
# Error in survdiff.fit(y, groups, strata.keep, rho) :
# There is only 1 group
# 有一些基因是没法进行分组的# 计算获得能够获得生存显著性及基因集合
prog_geneset = survGroup(maf = maf, top = 20, # 应用突变频率前20的基因集作为母集geneSetSize = 2, # 每2个基因作为signature计算的依据time = "OS.time", Status = "OS", verbose = FALSE)
prog_geneset
# Gene_combination P_value hr WT Mutant
# <char> <num> <num> <int> <int>
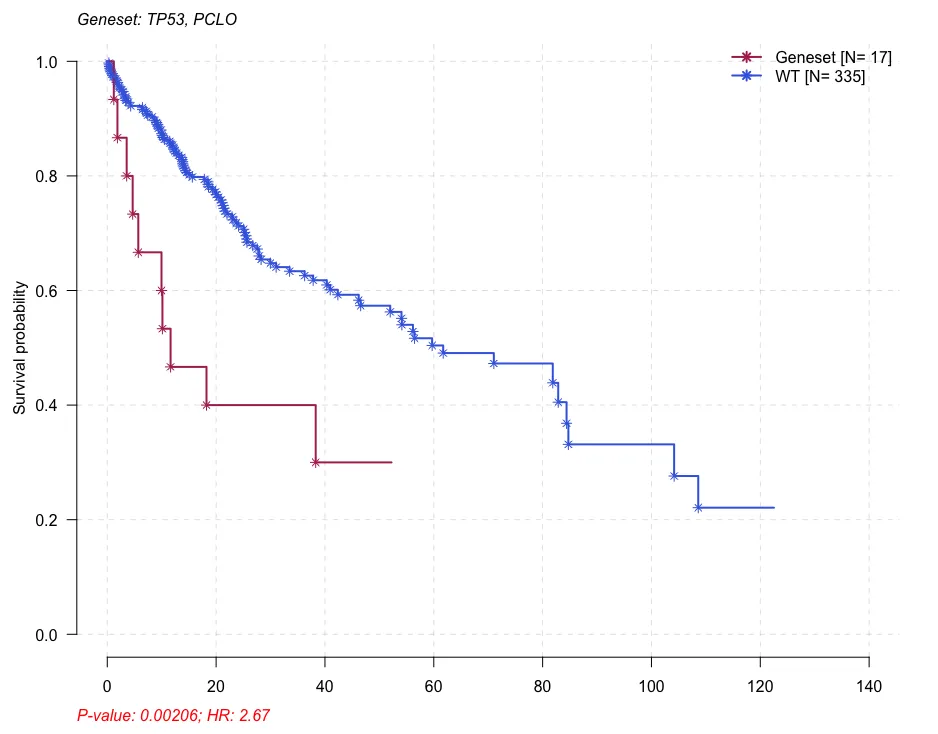
# 1: PCLO_GPR98 0.000168 5.580 346 6
# 2: PCLO_OBSCN 0.001350 3.940 344 8
# 3: TP53_PCLO 0.002060 2.670 335 17
# 4: MUC16_HMCN1 0.002610 3.630 346 6
# 5: TP53_FAT3 0.007420 2.460 338 14
# ---
# 105: CTNNB1_MUC16 0.950000 1.030 332 20
# 106: MUC16_RYR2 0.950000 0.964 341 11
# 107: ALB_ABCA13 0.996000 1.000 344 8
# 108: ARID1A_AXIN1 0.997000 1.000 347 5
# 109: TTN_XIRP2 0.998000 1.000 345 7# 指定基因进行分析
mafSurvGroup(maf = maf, geneSet = c("TP53", "PCLO"), time = "OS.time", Status = "OS")
12、临床表型与突变图谱的相关性分析
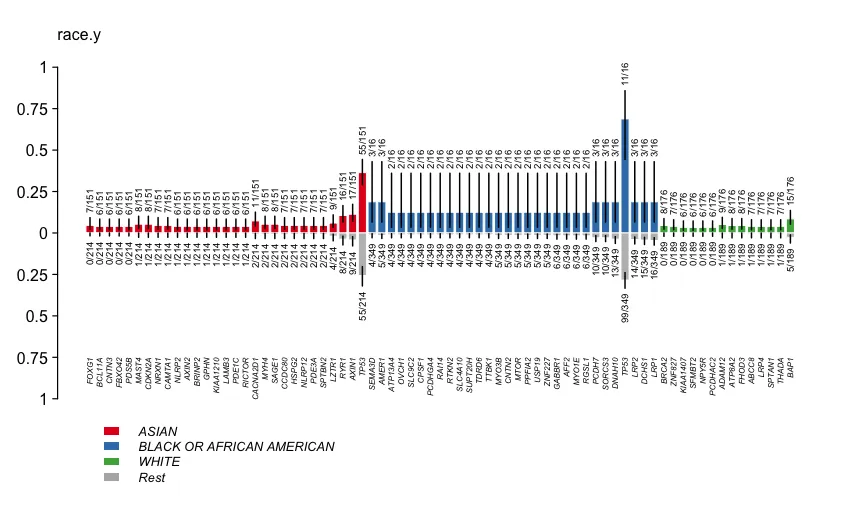
race = clinicalEnrichment(maf = maf, clinicalFeature = 'race.y')OS = clinicalEnrichment(maf = maf, clinicalFeature = 'OS')
#查看有统计学意义的突变类型
race$groupwise_comparision[p_value < 0.05]
# Hugo_Symbol Group1 Group2 n_mutated_group1 n_mutated_group2 p_value OR OR_low OR_high fdr
# <char> <char> <char> <char> <char> <num> <num> <num> <num> <num>
# 1: TP53 BLACK OR AFRICAN AMERICAN Rest 11 of 16 99 of 349 0.001285001 5.5255089 1.71654429 20.8205063 1
# 2: TP53 WHITE Rest 39 of 176 71 of 189 0.001414001 0.4740894 0.28942050 0.7687838 1
# 3: FOXG1 ASIAN Rest 7 of 151 0 of 214 0.001908274 Inf 2.09742173 Inf 1
# 4: SEMA3D BLACK OR AFRICAN AMERICAN Rest 3 of 16 4 of 349 0.002186009 19.3625522 2.57688879 127.7694190 1
# 5: CACNA2D1 ASIAN Rest 11 of 151 2 of 214 0.002373209 8.2837736 1.76888072 77.9776396 1
# ---
# 111: DIP2C ASIAN Rest 0 of 151 7 of 214 0.044474868 0.0000000 0.00000000 0.9691803 1
# 112: SCAPER ASIAN Rest 0 of 151 7 of 214 0.044474868 0.0000000 0.00000000 0.9691803 1
# 113: TRERF1 ASIAN Rest 0 of 151 7 of 214 0.044474868 0.0000000 0.00000000 0.9691803 1
# 114: LRP2 ASIAN Rest 3 of 151 14 of 214 0.045851136 0.2904066 0.05257406 1.0669070 1
# 115: LZTR1 ASIAN Rest 9 of 151 4 of 214 0.046441601 3.3165026 0.90431753 15.0216913 1#对有意义的临床表型-突变组合数据进行可视化
plotEnrichmentResults(enrich_res = race, pVal = 0.05, geneFontSize = 0.5, annoFontSize = 0.6)plotEnrichmentResults(enrich_res = OS, pVal = 0.05, geneFontSize = 0.5, annoFontSize = 0.6)
灰色代表的是未分类的信息。

13、药物互作
#查看药物-突变相关作用(潜在可靶药物)作用
drug = drugInteractions(maf = maf, fontSize = 0.75)#查看数据库中特定基因的可靶药物
drug_gene = drugInteractions(genes = "TP53", drugs = TRUE)
# Number of claimed drugs for given genes:
# Gene N
# <char> <int>
# 1: TP53 193
drug_gene
# Gene interaction_claim_source interaction_types drug_claim_name drug_claim_primary_name drug_name drug_chembl_id
# <char> <char> <char> <char> <char> <char> <char>
# 1: TP53 CKB CTX-1 CTX-1
# 2: TP53 CKB Gemcitabine Gemcitabine GEMCITABINE CHEMBL888
# 3: TP53 ClearityFoundationClinicalTrial GRANISETRON GRANISETRON GRANISETRON CHEMBL289469
# 4: TP53 CKB epirubicin epirubicin EPIRUBICIN CHEMBL417
# 5: TP53 NCI TOPICAL CORTICOSTEROIDS TOPICAL CORTICOSTEROIDS
# ---
# 189: TP53 CKB CEP-8983 CEP-8983
# 190: TP53 CKB Panitumumab Panitumumab PANITUMUMAB CHEMBL1201827
# 191: TP53 NCI STREPTONIGRIN STREPTONIGRIN STREPTONIGRIN CHEMBL11417
# 192: TP53 CKB Seliciclib Seliciclib SELICICLIB CHEMBL14762
# 193: TP53 CKB PD0166285 PD0166285
# PMIDs
# <char>
# 1: 26883273
# 2: 27167172,23520471,21389100,27815358,26228206
# 3:
# 4: 22903472
# 5: 12207591
# ---
# 189: 23428903
# 190: 28514312
# 191: 12027749
# 192: 22862161,26826118
# 193: 11719452
图表解释
纵轴(Y轴):展示了不同的药物靶向类别,如“TYROSINE KINASE”、“DNA REPAIR”、“CELL SURFACE”等。
横轴(X轴):显示了每个类别中基因的数量,范围从0到12。 类别和基因
每个类别旁边括号内的是该类别中的基因,例如:TYROSINE KINASE [TTN]:表示酪氨酸激酶类包含TTN基因。DNA REPAIR [TP53]:表示DNA修复类包含TP53基因。
数据解读
基因数量:条形的长度表示该类别中涉及的基因数量。图中的黑色条形突出显示了每个类别中的基因数量,有助于快速识别哪些类别含有较多的潜在药物靶点基因。
药物靶点类别:这些类别根据其在药物开发中的重要性和功能进行了组织,提供了潜在的药物作用靶点的概览。

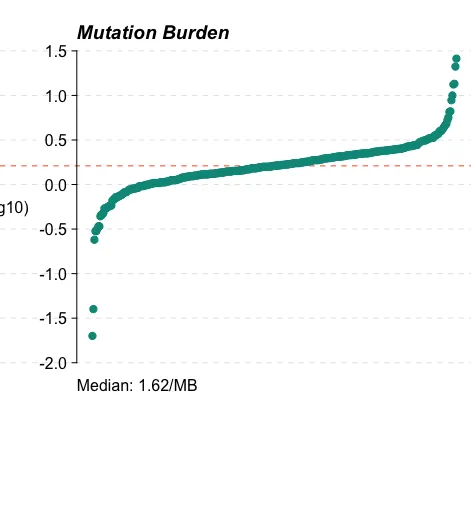
14、突变负荷分析
tmb(maf = maf)
# Tumor_Sample_Barcode total total_perMB total_perMB_log
# <fctr> <num> <num> <num>
# 1: TCGA-BC-A10X-01A-11D-A12Z-10 1 0.02 -1.6989700
# 2: TCGA-ED-A627-01A-12D-A30V-10 2 0.04 -1.3979400
# 3: TCGA-XR-A8TE-01A-11D-A35Z-10 12 0.24 -0.6197888
# 4: TCGA-5R-AA1D-01A-11D-A382-10 15 0.30 -0.5228787
# 5: TCGA-CC-A9FV-01A-11D-A36X-10 15 0.30 -0.5228787
# ---
# 361: TCGA-DD-AAC8-01A-11D-A40R-10 500 10.00 1.0000000
# 362: TCGA-DD-A1EE-01A-11D-A12Z-10 663 13.26 1.1225435
# 363: TCGA-CC-A7IH-01A-11D-A33K-10 675 13.50 1.1303338
# 364: TCGA-4R-AA8I-01A-11D-A382-10 1054 21.08 1.3238706
# 365: TCGA-UB-A7MB-01A-11D-A33Q-10 1294 25.88 1.4129643
参考资料:
1、Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018 Nov;28(11):1747-1756. doi: 10.1101/gr.239244.118 IF: 6.2 Q1 B2.
2、Maftools说明文档: https://bioconductor.org/packages/release/bioc/vignettes/maftools/inst/doc/maftools.html#1_Introduction
3、生信技能树: https://mp.weixin.qq.com/s/EHrJCEl8FL_3pdDxvuY7Tw https://mp.weixin.qq.com/s/Lx3ExJd6hg1sYMwEzugT2Q
4、生信菜鸟团:https://mp.weixin.qq.com/s/looYjwANerhPUMd7v0_5Fg
5、生信星球:https://mp.weixin.qq.com/s/z-8K95DJ-aIOdQMubqpMqQ
6、医学和生信笔记:https://mp.weixin.qq.com/s/1cR3Cnfd5Co9U3jIoIWJBA
7、聊生信:https://mp.weixin.qq.com/s/vKM-VMyFtLcW_VlnCkDngw
8、芒果师兄聊生信:https://mp.weixin.qq.com/s/V6lce2wCR2asJCEewcvwqA
9、灵活胖子的科研进步之路:https://mp.weixin.qq.com/s/6vllQmjsem8jEZgBsrayuQ
10、生信碱移:https://mp.weixin.qq.com/s/pBDue6hOf0DrTZMN0rB2sA
11、小明的数据分析笔记本:https://mp.weixin.qq.com/s/Qi9rkGAV0r__tvnIXoLSYQ
12、Bio小菜鸟:https://mp.weixin.qq.com/s/vC3pBavzLOJrtaiRIDB2Ag
注:若对内容有疑惑或者有发现明确错误的朋友,请联系后台(欢迎交流)。更多内容可关注公众号:生信方舟
- END -
这篇关于maftools癌症体细胞变异(突变)分析工具学习的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!







