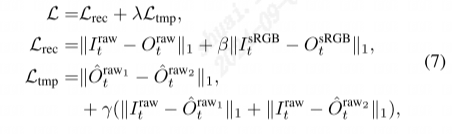本文主要是介绍茄科四个参考基因组-文献精读41,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
Multiple independent losses of the biosynthetic pathway for two tropane alkaloids in the Solanaceae family
茄科植物中两种莨菪烷生物合成途径的多次独立丧失

摘要
东莨菪碱和莨菪碱(HS)是两种具有重要药用价值的莨菪烷生物碱,它们存在于茄科家族中多个关系较远的谱系中。在本研究中,我们测序了来自这些谱系的三种代表性HS产生物种的基因组,以及一种不产生HS的物种。我们的分析揭示了这三种HS产生物种中共同负责HS合成的生物合成途径。我们观察到在茄科家族中两类物种中与HS合成相关的基因高度共线性。通过在关键位点引入功能获得和功能丧失突变,我们分别验证了两类物种中与HS合成相关的关键基因功能的减少/丧失或重新激活。这些发现表明,自其在祖先谱系中起源以来,HS生物合成途径经历了独立且反复的丧失。我们的结果为未来在茄科作物中人工工程HS生物合成提供了潜在的应用前景。
引言
植物中的次级代谢物是强大的进化选择的结果。令人瞩目的是,完全相同的植物次级代谢物可以在关系较远的谱系中被发现,这表明自然选择通过生物和非生物胁迫(包括防御病原微生物和食草动物、与授粉者的相互作用、以及对抗干旱、紫外线辐射和霜冻的保护)多次作用,生成这些代谢物。另一种解释是,这些代谢途径可能起源于所有相关谱系的最近共同祖先,并在关系较远的谱系中保留了下来,但由于不利的选择压力或在分化过程中发生的遗传漂变,可能在某些关系较近的谱系中丧失。
莨菪烷生物碱是植物生物碱多样化的吡咯啉类成员,其化学结构中包含一个8-甲基8-氮杂双环[3.2.1]辛烷(莨菪碱)环。目前,已经在包括茄科、旋花科、红树科和古柯科等多个植物家族中鉴定出超过300种莨菪烷生物碱。这些莨菪烷生物碱可以根据其生物合成和结构特征分为几个主要类别。例如,古柯碱来源于古柯科,东莨菪碱和莨菪碱(HS)在茄科中产生,而卡立净则从旋花科中分离出来。最近的研究表明,存在于古柯科和茄科中的这些莨菪烷生物碱是独立进化的,但在功能上表现出趋同,这通过酶的特性分析得以证实。在茄科家族中,HS的生产可见于四个不同的系统发育和地理谱系中。Datureae部族起源于南美洲,而Mandragorinae亚科和Hyoscyaminae亚科(包括之前归类为姐妹群Lyciinae的Atropa)则分布在青藏高原及其邻近地区,Anthocercideae部族则出现在澳大利亚。这些生物碱具有多种应用,常用于神经毒剂中毒、帕金森病和神经肌肉疾病的治疗,是多种有效药物生产的基础。此外,消旋HS是一种世界卫生组织认可的有效、安全且价格低廉的药物。然而,从相应植物中分离出的HS的全球供应仍然是一个挑战。因此,理解HS生产的基因进化,并明确负责这一生产的特定基因和突变,可能有助于在未来通过工程化途径生成这两种具有生物活性和经济价值的产品。
一个多世纪以来,已经利用多种HS生产物种来揭示HS生物合成途径的每一步。目前,整个HS生物合成途径已在颠茄中完全表征。据推测,共有12种酶参与HS的生物合成,该过程从多胺腐胺(1)开始,腐胺由两个初始氨基酸前体鸟氨酸或精氨酸衍生而来。1通过腐胺甲基转移酶(PMT)的作用被甲基化,生成N-甲基腐胺(2),随后通过N-甲基腐胺氧化酶(MPO)被氧化,生成4-甲基氨基丁醛(3)。3随后经历自发环化,生成N-甲基吡咯啉阳离子(4),通过聚酮合酶(PYKS)和细胞色素P450(CYP82M3)酶介导的途径转化为托品酮(5)。托品酮还原酶I(TRI)随后将5转化为托品(6),这是HS合成的初始起始材料。芳香族氨基酸氨基转移酶(ArAT4)催化苯丙氨酸(7)转化为苯丙酮酸(8)。苯丙酮酸还原酶(PPAR)将8还原为苯乳酸(9),随后由UDP糖基转移酶(UGT1)转化为苯乳糖苷(10),这是生成莨菪花碱(11)的酰基供体。莨菪花碱(11)通过莨菪花碱合酶(LS)催化的步骤与6和10结合生成,随后通过P450介导的(CYP80F1)重排生成莨菪碱醛(12)。莨菪碱脱氢酶(HDH)将12转化为莨菪碱(13),随后通过莨菪碱6-羟化酶(H6H)催化的两步环氧化反应转化为东莨菪碱(14)。在HS生物合成所需的12个步骤中,从TRI作用开始的最后五个步骤仅专用于HS的生产。相对而言,前四种酶及其相应的产物参与多种代谢物的合成,包括古柯碱和卡立净。值得注意的是,TRI生成的托品(6)是HS途径中的一个关键中间代谢物。此外,LS是另一个关键基因,对于启动专门的HS生物合成至关重要。
上述研究进展引发了许多科学问题。特别是,尚不清楚四个HS生产谱系是否使用相同的生物合成基因来生成HS。如果这些谱系遵循相同的HS合成遗传途径,那么HS生物合成的独立起源可能需要多个关键基因的趋同功能。或者,这些次级代谢产物可能起源于一个祖先途径,而这一途径在关系较近的谱系中丧失。因此,我们试图评估这些在茄科中系统发育距离较远的植物中分布零散的药用莨菪烷生物碱HS生物合成的进化选择。
在本研究中,我们测序了来自茄科家族中三个关系较远谱系(分别为Datureae、Hyoscyaminae和Mandragorinae)中生产HS的代表物种,包括山莨菪、木本曼陀罗和茄参。此外,我们为非HS生产物种——与Hyoscyameae关系密切的中国枸杞——产生了高质量的基因组组装。此外,我们还获得了13个代表多个非HS生产谱系的非HS生产物种的高质量基因组用于比较。我们的主要目标是评估这些茄科家族中关系较远的谱系中HS生物合成的进化历史。我们评估了基因组结构,搜索了代表所有HS相关基因在每个合成步骤中保守进化的同线性块,并通过在两类物种中引入功能获得和功能丧失突变,测试了HS生物合成酶关键位点的存在。这些基因和关键突变的表征对于未来在广泛种植的茄科作物(包括番茄和马铃薯)中工程化HS途径以有效获取这两种代谢产物具有重要意义。
| Category | 山莨菪 | 中国枸杞 | 木本曼陀罗 | 茄参 |
|---|---|---|---|---|
| Sequencing | ||||
| Platform | Nanopore | PacBio | PacBio | PacBio |
| Genome-sequencing depth (X) | 105.77 | 35.57 (valid) | 20.09 (valid) | 33 (valid) |
| Assembly | ||||
| Estimated genome size (Mb) | 1198 | 1408 | 1517 | 756 |
| Assembled genome size (Mb) | 1249 | 1538 | 1548 | 712 |
| N50 of scaffolds (bp) | 49,959,515 | 132,783,878 | 121,782,173 | 28,080,016 |
| No. of contigs | 205 | 1,406 | 1,398 | 808 |
| N50 of contigs (bp) | 23,808,256 | 2,994,494 | 7,701,694 | 25,262,060 |
| GC content of the genome (%) | 37.05 | 37.66 | 35.22 | 35.18 |
| Anchored to chromosome (%) | 97.47 | 98.53 | 97.40 | 94.67 |
| Complete BUSCOs (%) | 98.10 | 94.90 | 98.10 | 98.30 |
| Annotation | ||||
| Percentage of repeat sequences (%) | 65.68 | 70.22 | 79.0 | 70.11 |
| LTR rate (%) | 43.07 | 44.48 | 58.37 | 36.62 |
| No. of predicted protein-coding genes | 46,606 | 54,946 | 32,347 | 29,193 |
| Average gene length (bp) | 4846.05 | 3775.38 | 3880.88 | 5447.13 |
| Average CDS length (bp) | 1134.68 | 1017.84 | 1128.02 | 1282.79 |
| Mean exon/intron length (bp) | 216.06/872.93 | 236.46/834.45 | 230.10/705.45 | 222.87/875.65 |
| Mean exon number per gene | 5.25 | 4.30 | 4.90 | 5.76 |

a. 显示了茄参、山莨菪、木本曼陀罗 和 中国枸杞 的基因组形态和 Circos 图。不同的轨道(从外向内)依次表示:(I)基因密度;(II)鸟嘌呤-胞嘧啶(GC)含量;(III)LTR-Gypsy 转座子密度;(IV)LTR-Copia 转座子密度。 b. 同义置换水平(Ks)在共线性同源基因中的分布情况。 c. 比较 山莨菪 与 木本曼陀罗 以及 茄参 之间的共线性点图。 d. 12 个物种的系统发育树及基因家族的进化。节点旁的黑色数值表示该特定节点的估计分歧时间(MYA,百万年前)。基因家族扩展和收缩事件的数量(p 值≦ 0.01)分别以红色和蓝色表示。
我们通过多种方法评估了基因组组装的质量。超过98.78%的 Illumina 短读段准确地映射到四个基因组上。测序的转录组数据也表现出较高的映射率,范围从81.10%到97.37%(补充表8)。组装的转录本被映射到基因组上,超过70.25%的转录本长度超过了各物种每条染色体的一半(补充数据1)。进行了 BUSCO 分析,发现三种 HS 产生物种中超过98%的 BUSCOs 可以被完全检索到,而在非HS产生物种 中国枸杞 中则获得了95%的完整 BUSCO 分数(补充表9)。这些结果确立了本研究所描述的四个基因组的高准确性、连续性和全面性。
基因组注释
结合 de novo、同源性和基于转录组的方法,预测了 山莨菪、中国枸杞、木本曼陀罗 和 茄参 基因组中的46,606、54,946、32,347 和29,193 个蛋白质编码基因(表1,补充表10-14,补充图4)。利用 BUSCO 对四个基因集中基因组注释的完整性进行了评估,分别为 山莨菪(94.9%)、中国枸杞(92.2%)、木本曼陀罗(93.8%)和 茄参(93.1%)(补充表15)。在 山莨菪、中国枸杞、木本曼陀罗 和 茄参 中,大约96.0%、92.0%、95.9% 和 98.56% 的蛋白质编码基因成功地使用至少一个数据库(包括 Swiss-Prot、KEGG、InterPro、Pfam、GO、NR 和 COG)进行了注释(补充表16)。与其他三个物种相比,预测到的重复元件在 木本曼陀罗 的基因组中所占比例显著提高(分别为65.68%、70.22% 和 70.11% 相比于 79.0%;补充表17)。在所有四个基因组中,转座元件(TEs),即被认为是被子植物基因组中最常见的重复形式,是最丰富的重复子类型(补充表18)。在 TEs 中,长末端重复序列(LTRs)是最丰富的,其中 木本曼陀罗 含有最高的 LTR 含量(58.37%),而 茄参 的 LTR 含量最低(36.62%)(补充表18)。此外,我们在 山莨菪、中国枸杞、木本曼陀罗 和 茄参 的基因组中分别分类了 1,752、2,884、2,325 和 2,727 个转录因子(TF)或转录调节因子(TR)(补充表19)。在四个物种中都鉴定出了多种非编码 RNA(ncRNA)基因(补充表20)。
基因组进化
我们考察了茄科家族内三种系统发育上无关的 HS 产生物种的多倍化历史。对于每个基因组,我们利用共线性同源基因确定了每个同义位点的同义置换分布(Ks)。我们在 山莨菪 中观察到一个约为 0.5 的小 Ks 峰,它出现在所有其他茄科物种中,对应于该家族中共有的全基因组三倍化事件。此外,我们在 山莨菪 和 茄参 中分别检测到约为 0.15 和 0.20 的显著的近期 Ks 峰,表明可能存在谱系特异的近期多倍化事件。
为了阐明四个物种的多倍化历史,我们利用葡萄(Vitis vinifera)及其他茄科物种作为参考物种进行比较基因组评估。我们检测到 山莨菪 和葡萄之间的共线性深度比为6:1,与 山莨菪 和 木本曼陀罗、中国枸杞 以及 Solanum tuberosum 之间的比率为 2:1。同样,我们在 茄参 与 中国枸杞 以及 茄参 与 S. lycopersicum 的比较中表征了2:1的共线性深度比,并在 茄参 和 S. melongena 以及 C. annuum 之间分别检测到了2:1的深度比。这些发现证实了 山莨菪 和 茄参 在与其他茄科物种共同的祖先全基因组三倍化(WGT)事件后,也经历了谱系特异的全基因组重复(WGD)事件。山莨菪 和 茄参 之间的共线性点图表明,每个物种中的每个片段都可以与另一个物种中的两个最相关的共线性片段相匹配,这验证了这些物种之间的近期 WGD 事件并不共享。
我们选择了12个物种(Oryza sativa、Cucumis sativus、V. vinifera、Mimulus guttatus、Petunia axillaris、Nicotiana attenuata、中国枸杞、山莨菪、茄参、木本曼陀罗、S. tuberosum 和 Solanum melongena)进行基因家族构建。大约404,865个基因被分为26,783个基因家族,并表征了220个单拷贝基因家族。根据这些单拷贝基因,推断了系统发育树和分歧时间。我们的研究结果表明,P. axillaris 在茄科家族中经历了最早的分歧,约发生在 27.33 百万年前(Mya)。木本曼陀罗 是茄属物种的姐妹群,并且与 茄参、山莨菪 和 中国枸杞 密切相关,这些物种共同构成了一个分歧时间为约21.17 Mya 的谱系。
我们使用CAFÉ对基因家族扩展和收缩进行了检查。在 山莨菪、中国枸杞、木本曼陀罗 和 茄参 中分别识别出了 8,629/1085、4,592/620、1,856/2534 和 4,490/533 个扩展和独特的基因家族。我们发现,通过对三个 HS 生产物种(山莨菪、木本曼陀罗 和 茄参)的 GO 分析,扩展和独特基因主要涉及杂环合成过程、血红素转运蛋白活性、ADP结合、对刺激的反应和蛋白质丝氨酸/苏氨酸激酶活性;而在非 HS 生产物种 中国枸杞 中,通过 GO 分析,扩展和独特基因主要涉及定向定位物质代谢过程和核酸结合。
三种 HS 合成物种具有共同的 HS 生物合成途径
鉴于 山莨菪、木本曼陀罗 和 茄参 都能生成 HS,可能是它们采用了一个保守的、具有共同进化历史的途径来生产这些化合物。为了进一步评估这一点,我们进行了比较基因组分析,旨在通过结合 BLAST 搜索和 HMM 方法并参考与 山莨菪 同系的颠茄中功能验证的基因序列,分类出这些物种中负责 HS 合成的基因序列。我们手动修正了自动注释中的错误,并将所有同源基因分类为12个酶家族,序列一致性高(>80%)、错配率低,且三个物种与颠茄之间的序列具有相同的结构域。我们还将这些基因与以往研究中功能验证的三种产生 HS 的物种中的基因进行了比较,发现这些基因在序列相似性和基因表达模式方面具有高度的保守性,这表明这些物种应共享一个共同的 HS 生物合成途径。为了更深入了解这些基因在植物中 HS 生物合成中的功能,我们从 HS 生物合成途径中选择了四个关键基因,包括 TRI、LS、HDH 和 H6H,在 山莨菪 中使用初步的病毒诱导基因沉默(VIGS)技术进行验证。实验结果表明,抑制 TRI、LS 和 H6H 基因的表达导致相应产品(即莨菪碱、莨菪花碱、东莨菪碱和莨菪碱)的减少。因此,我们得出结论,这些候选基因是 HS 生产物种中 HS 生物合成所必需的基因。我们在每个 HS 生产物种中检测到了包含12种酶的可能基因簇,并确定在 山莨菪 和 木本曼陀罗 中,只有 CYP82M3 和 TRI 聚集在一起,而其余基因则分布在不同的基因组位点。我们进一步检测了在三种 HS 生产物种(叶、根、次生根和茎)的不同组织中鉴定基因的表达水平,并发现所有候选基因都在这三种物种的根和叶中表达。值得注意的是,这些基因在 山莨菪 的次生根中的表达尤其高,这有助于从这些物种的根中分离出高浓度的 HS。

a. 概览显示了HS生物合成途径,以及 山莨菪(Ata)、木本曼陀罗(Bar)和 茄参(Mca)中候选基因的表达谱热图。图中显示了 木本曼陀罗 中四个序列相似度超过90%的串联重复TRI基因。缩写说明,sec_root:次生根。 b. 系统发育树为茄科15个选定物种构建,使用旋花科的三裂旋花和琉璃苣作为外部参照(外群)。红色条代表 山莨菪 和 茄参 中的全基因组重复(WGD)事件(左图)。通过17个物种的微共线性分析识别了TRI基因区域。矩形表示注释基因,方向为反向链(绿色)和正向链(蓝色)。山莨菪、木本曼陀罗 和 茄参 的TRI基因名称标记在基因块上方。共线性TRI基因之间的连线以红色突出显示,CYP82M3基因以蓝色突出显示。灰色连线表示候选物种间的基因共线性。红色虚线表示 山莨菪 和茄子中不完整TRI基因(伪基因)的假设共线性(右图)。源数据作为源数据文件提供。
茄科家族中HS合成基因的保守共线性块
保留的共线性块中存在与特定性状相关的关键基因,而这些基因在缺乏该性状的近缘物种中仍然存在,这提供了失去祖先遗传途径的证据。我们在三个系统发育上关系较远的HS生成物种中,识别出了12个HS合成基因中的11个(包括PMT、MPO、PYKS、CYP82M3、AT4、UGT1、TRI、LS、CYPB80F1、HDH和H6H基因家族)的高度保守块。此外,除了HDH外,这11个基因中的10个在至少一个非HS生成的茄科物种中拥有共线性基因及同源的完整或不完整的基因。
例如,TRI基因的下游关键基因的共线性块在所有茄科物种中都被识别出来,其中包含了所有物种中串联重复或不完整的TRI直系同源片段。HS生成物种 山莨菪 的2号染色体中包含AtTRI,与18号染色体表现出高度的物种内共线性(补充图23),其中包含不完整的同源TRI。我们计算了这两条染色体之间共线性基因对的同义置换率(Ks)以确定该重复事件的时间尺度,结果得到的时间为13.41百万年前(95%置信区间:7.93-18.89百万年前)(补充表22)。这个时间与 山莨菪 中的物种特异性WGD事件(图1b)很好地吻合。该物种的TRI基因起源于特定WGD事件之前。TRI基因树与茄科家族的物种树排列相似(补充图24、28、29),表明TRI的进化历史与茄科物种之间存在关系。有趣的是,在对PPAR进行微共线性分析时,我们无法直接在 木本曼陀罗、山莨菪 和 茄参 以及非HS生成物种(例如 Petunia inflata、P. axillaris 和 中国枸杞)之间识别出共线性块(补充图30a)。然而,通过全基因组基因家族分析,我们在这些物种中验证了候选直系同源基因(补充图30b),这表明在谱系分化过程中可能发生了基因易位和共线性侵蚀。这些因素可能是导致共线性差异的原因。HS合成基因的高度保守共线性块及其系统发育关系表明,HS合成途径可能起源于所有茄科谱系的共同祖先。

a. LS基因在17个选定物种中的微共线性分析。矩形表示注释的基因,方向为反向链(绿色)和正向链(蓝色)。山莨菪、木本曼陀罗 和 茄参 的LS基因名称标记在基因块上方。共线性LS基因之间的连线以红色突出显示。灰色连线表示候选物种间的基因共线性。 b. 长筒蓝悬铃果 中LS残基的位置,黑色圆角矩形表示外显子。 c. 从莨菪花碱和莨菪碱醛分支的生物合成途径。 d. CYP80F1基因在P. inflata、辣椒、木本曼陀罗、山莨菪和茄参之间的共线性评估。山莨菪、木本曼陀罗 和 茄参 的CYP80F1基因名称标记在基因块上方。黑色虚线矩形表示CYP80F1残基的位置,虚线框中的绿色圆角矩形表示外显子。连线表示具有共线性的基因,粉红色连线表示 P. inflata、木本曼陀罗、山莨菪 和 茄参 之间CYP80F1基因的共线性关系。粉红色虚线表示三个辣椒物种中不完整CYP80F1基因(伪基因)的假定共线性。

系统发育树包括生产HS的物种(红色)和不生产HS的茄科物种(蓝色),其中 I. nil 和 I. triloba 用作外群。12个HS基因的缺失、存在或片段(伪基因)分别用白色、黑色和黑色三角形框表示。星号表示HS生物合成途径的独立丧失。星号表示为本研究测序的物种。托品环形成是HS生物合成途径的上游基因;PLA葡萄糖苷合成是HS生物合成途径的分支基因;药用HS合成是HS生物合成途径的下游基因。
与展示共线性保守性的其他HS合成基因相比,我们发现三个下游HS特异性基因LS、CYP80F1和HDH在不生产HS的物种中表现出不完整的基因结构,类似于伪基因。例如,在一个不生产HS的物种 长筒蓝悬铃果 中,LS基因的前两个外显子发生了进化变化,生成了伪基因。类似地,在不生产HS的辣椒物种中识别出了CYP80F1基因的共线性伪基因。虽然我们无法在不生产HS的茄科物种中找到HDH残基,但我们在同属茄目、关系密切的旋花科物种中确认了其共线性块和完整的基因结构。这表明HDH基因在两个家族分化之前具有祖先起源。这些基因在不生产HS的物种中缺乏完整的基因结构可能与伪基因化有关,导致外显子的丧失。或者,这些基因可能存在于未采样的不生产HS的物种中,随着高质量基因组的广泛可用性,未来可能进行研究。这些结果强调了三个系统发育关系较远的HS生产物种中HS合成基因的显著共线性,以及它们在不生产HS的物种和关系密切的家族中的部分或完全保留。此外,三种HS生产物种中每个位点的同源基因被确定为并系群,而不是在趋同进化情况下预期的单系群。因此,不生产HS的物种中共线性区域内HS合成下游基因的伪基因化和丧失导致了HS生物合成在最近共同起源后的多次丧失。
TRI关键基因的功能活性实验测试
TRI基因对于生产HS合成途径中的关键中间代谢物托品(6)至关重要。我们以TRI作为代表基因,旨在检验HS合成基因自祖先起源以来在不生产HS的物种中可能经历了功能衰减或丧失的假设。与其他HS合成基因相比,TRI在不生产HS的物种中表现出多样化的模式,范围从完整形式的保存到演变为伪基因或完全缺失。这种独特的变异使TRI成为在多个位点进行功能丧失和功能获得实验的最佳候选基因,结合在生产HS和不生产HS的物种中的相互突变实验。通过调节TRI,我们可以深入了解这些基因改变的功能重要性,并探索HS合成基因在生物合成途径中的作用。这种方法有望阐明不生产HS的物种在进化历史过程中HS生物合成丧失或减少的机制。
我们检查了来自17个茄科物种的TRI序列,并鉴定出高度的序列同一性,三种生产HS的物种之间的相似性为90%,所有物种的相似性为82%。我们鉴定出五个可变位点(109、155、167、201和243),它们对于同源TRI基因的限制至关重要。虽然这些位点在生产HS的物种中是保守的,但由于松散的进化,它们在不生产HS的物种中表现出变异。我们模拟了 木本曼陀罗 BaTRI蛋白的三级结构,并通过分子对接识别出包含BaTRI、NADPH和5的三元复合物。我们发现Val109和Val167形成了一个能够结合托品酮的疏水口袋。Val109与5之间的距离估计为3.6-3.9Å,而Val167与5之间的距离为3.8Å。这两个残基通过疏水力在将5置于优选的催化构象中起到稳定作用。此外,祖先序列重建表明在109和167两个位置上的Val的概率超过99%。

a. 比较了参与HS生产和非HS生产植物物种中的TRI关键氨基酸(五个TRI分别来自茄参 (McTRI)、木本曼陀罗 (BaTRI)、山莨菪 (AtTRI)、中国枸杞 (LcTRI) 和 S. tuberosum (StTRI))。星号表示通过本研究确认的酶。蓝色高亮部分表示催化四联体,橙色高亮部分表示关键位点。值得注意的是,位点109和167也是托品酮的结合位点。位点在BaTRI蛋白中的相对位置用表示。序列中的点表示省略的位点。 b. BaTRI的三级结构。两个紫色球棒模型表示功能验证的关键位点。 c. 提取离子色谱图(EIC)显示了七种纯化的重组TRI(来自BaTRI、AtTRI、McTRI、LcTRI、StTRI、StTRIA109V、L167V 和 StTRIA109V、V155L、L167V、A201G)与托品酮作为底物的体外活性。 d. 五种野生型TRI(d)和五种突变型TRI(e)的还原催化效率(Kcat/Km值)的评估。这些TRI对托品酮进行NADPH依赖性还原反应的原始Michaelis-Menten曲线在补充图33中显示。误差条以n=3的生物独立实验的平均值±SD表示。d 和 e 中不同的数字表示在双尾Student’s t检验中p < 0.001 () 时值显著不同。ND表示未检测到。I (p = 0.0009),II* (p = 0.0013),III** (p = 0.0044),IV** (p = 0.0002) 和 V** (p = 0.0013) 表示与AtTRI和LcTRI、BaTRI和LcTRI、McTRI和LcTRI、BaTRIV109A和BaTRI、BaTRIV167L和BaTRI相比值显著不同。 f. 提取离子色谱图显示了通过共表达AbUGT1基因(三个LS基因来自山莨菪 (AtLS)、木本曼陀罗 (BaLS) 和 A. belladonna (AbLS))在烟草叶片中催化托品(6)和苯乳糖苷(10)转化为莨菪花碱(11)的体内活性。苯乳酸和托品作为底物通过共渗入烟草叶片中,莨菪碱和莨菪花碱的提取离子色谱图以浅蓝色表示。源数据作为源数据文件提供。
我们利用三种HS生产物种(山莨菪、木本曼陀罗 和 茄参)和两种缺少已识别位点的非HS生产物种(中国枸杞 和 S. tuberosum),研究了TRI蛋白催化的NADPH依赖性托品酮(5)还原反应。通过超高效液相色谱-质谱(UPLC-MS)验证了TRI催化的还原反应。与托品(6)的母离子(m/z = 142.12)对应的MS光谱比较,发现 BaTRI(木本曼陀罗)、AtTRI(山莨菪)、McTRI(茄参)和 LcTRI(中国枸杞)样品在保留时间为7.12分钟时有一个峰,产生的片段保留时间与使用托品(6)标准物质鉴定的片段相同。在 StTRI(S. tuberosum)中未检测到托品(6)峰。各物种的TRI活性与其原始物种中记录的托品(6)浓度相匹配。
我们为TRI酶及其托品酮(5)底物生成了Michaelis-Menten曲线,以确定Km和Vmax值,并使用这些值推导出Kcat和催化效率(Kcat/Km)值。我们的研究发现,LcTRI 对托品酮(5)的亲和力(Km = 2.91 ± 0.14 mM)比 BaTRI 和 AtTRI(分别为Km = 0.31 ± 0.03 mM 和Km = 0.21 ± 0.01 mM)低10倍。然而,在研究HS生产物种 茄参 的 McTRI 时出现了一个有趣的结果。McTRI 显示出显著增加的对托品酮的亲和力,Km值为0.09 ± 0.01 mM,表现出卓越的催化效率(Kcat/Km值为61.46 ± 5.76)。这些值代表了迄今为止在任何直系同源基因中报告的最大亲和力,表明 McTRI 是未来HS及其他莨菪烷生物碱代谢工程的有前途的候选者。此外,AtTRI、BaTRI 和 McTRI 对托品酮(5)的催化效率(Kcat/Km值分别为1.55、5.60 和61.46)比 LcTRI(0.24)显著更快更强(p = 0.0009、p = 0.0013 和 p = 0.0044,p < 0.01,t检验)。相反,来自土豆的 StTRI 没有表现出任何活性。
为了验证我们关于非HS生产物种中TRI基因的特定位点可能由于松散进化而表现出活性降低或丧失的假设,我们对非HS生产的土豆和HS生产的 木本曼陀罗 中的 StTRI 和 BaTRI 分别进行了功能获得和功能丧失的突变实验。通过序列分析和全面的文献回顾,确定 BaTRI 蛋白中的 Val109、Leu155、Val167 和 Gly201 残基对于维持高活性至关重要。随后,我们在 StTRI 和 BaTRI 中进行了关键氨基酸替换,并检查了它们的酶动力学,包括在功能获得突变中 StTRI A109V、L167V 和 StTRI A109V、V155L、L167V、A201G,以及在功能丧失突变中 BaTRIV109A、BaTRIV167L、BaTRIV109A、V167L。突变体 StTRI A109V、L167V 和 StTRI A109V、V155L、L167V、A201G 显示出Kcat/Km值显著增加,表明对托品酮(5)转化为托品(6)的催化能力增强。相比之下,BaTRIV109A 和 BaTRIV167L 的Kcat/Km值相对于 BaTRI 显著降低(p = 0.0002 和 p = 0.0013,p < 0.01,t检验)。有趣的是,BaTRIV109A、V167L 完全丧失了将托品酮(5)转化为托品(6)的能力。这些发现提供了证据,表明在系统发育关系较远的HS生产物种中,关键的TRI基因保持了其祖先的活性功能。相反,非HS生产物种,尽管拥有完整的TRI基因(如在土豆中观察到的那样),由于松散进化可能经历了功能的降低甚至完全丧失。
关键下游HS合成基因的功能丧失
关键下游HS合成基因的功能丧失可能通过与功能终止、基因伪基因化和非HS生产物种中基因丧失相关的保守进化过程发生,而这些基因在HS生产物种中保留了其祖先功能。例如,我们确认了LS基因在
HS生产物种中催化托品(6)和苯乳糖苷(10)转化为莨菪花碱(11)的活性。然而,我们观察到某些非HS生产物种仍然在HS合成途径的下游具有共线性基因,即PPAR、CYP80F1、HDH和H6H。因此,研究这些非HS生产物种中这些基因是否保留了酶活性是值得的。
为了研究PPAR的酶功能,我们通过体外酶实验描述了来自 木本曼陀罗、茄参 和 P. inflata 的PPAR功能。与之前的研究一致,所有检测的PPARs都将苯丙酮酸(8)转化为苯乳酸(9)。关于CYP80F1同源基因,我们克隆了这些基因并在烟草叶片中瞬时表达。结果表明,与其他HS生产物种相比,P. inflata(PiCYP80F1)的CYP80F1基因缺乏完整的功能域。然后,我们通过与 AbHDH、AtHDH、BaHDH、McHDH、ItHDH(I. triloba)和 InHDH(I. nil)瞬时共表达在烟草叶片中对四种CYP80F1同源基因(山莨菪(AtCYP80F1)、木本曼陀罗(BaCYP80F1)、茄参(McCYP80F1)和 A. belladonna(AbCYP80F1))的相对酶活性进行了检查,这些基因分别从对应的物种中克隆,用于将莨菪花碱(11)转化为莨菪碱(13)。在注入莨菪花碱(11)后,我们测量了所有表达相应构建体的烟草叶片中的莨菪花碱(11)和莨菪碱(13),表明了所有检测酶的活性。H6H基因的生物信息学评估和体外酶活性实验表明 茄参(McH6H)和 中国枸杞(LcH6H)的H6H基因具有酶活性。先前的研究报道了山莨菪 和 木本曼陀罗 的H6H基因催化了生产东莨菪碱(14)的两步反应。因此,所有这些结果表明,HS合成途径可能起源于所有茄科谱系的共同祖先,这些基因在某些非HS生产物种中的丧失或伪基因化导致了HS在茄科家族中的有限分布。

a. 提取离子色谱图(EIC)展示了 BaPPAR、McPPAR 和 PiPPAR 在体外以苯丙酮酸为底物的活性。 b. 提取离子色谱图显示了四种 CYP80F1 基因(分别来自 山莨菪 (AtCYP80F1)、木本曼陀罗 (BaCYP80F1)、茄参 (McCYP80F1) 和 A. belladonna (AbCYP80F1))的体内活性,这些基因通过共表达 AbHDH、AtHDH、BaHDH、McHDH、ItHDH (I. triloba) 和 InHDH (I. nil) 基因,在烟草叶片中将莨菪花碱转化为莨菪碱。莨菪花碱通过共渗入烟草叶片中作为底物。 c. 提取离子色谱图(EIC)展示了 McH6H 和 LcH6H 在体外以莨菪碱或东莨菪碱为底物的活性。源数据作为源数据文件提供。
讨论
我们成功编译了四个茄科物种的高质量基因组,其中包括三种生产HS的物种(山莨菪、木本曼陀罗 和 茄参)以及一种不生产HS的物种(中国枸杞)。结合先前报道的非HS生产物种的基因组,我们发现了每个基因组的进化历史,并揭示出 山莨菪 和 茄参 发生了额外的谱系特异性全基因组重复(WGD)事件,与其他物种共享的茄科家族三倍化事件相比。虽然 中国枸杞 没有经历谱系特异性WGD事件,但我们观察到其基因组内存在高水平的基因重复。这些重复可能源于该物种保留了祖先WGT事件中的大量基因,或可能涉及需要在未来进一步研究的其他技术因素。
我们的结果表明,茄科家族中三个系统发育关系较远的物种可能遵循相同的HS生物合成途径。此外,我们通过VIGS和生化方法揭示了来自HS生产高山物种的 TRI、LS、PPAR、CYP80F1、HDH 和 H6H 的作用。上述体外方法有效地验证了与HS相关基因的功能。此外,在HS生产物种中过表达 TRI 基因可以增强托品(6)的生产。相反,我们旨在降低 山莨菪 中 TRI 基因表达的VIGS实验导致了托品(6)浓度的降低。这些互补实验加强了 TRI 基因在HS生物合成中的关键作用,并提供了持续一致的证据。值得注意的是,构建的物种树支持了先前研究的发现,即三个HS生产物种(代表三个不同群体)在系统发育上是分散的,并不形成单系群。HS生物合成途径的这种系统发育分布表明,它可能是通过趋同进化或在祖先起源后通过多个独立丧失产生的,并在系统发育上无关联的物种中表现为共线性块和共线性基因。我们注释了共线性块,并在三个非单系的HS生产物种中表征了每个HS合成阶段的同源基因。此外,对于所有12个HS合成基因,我们在至少一个非HS生产物种中识别出共线性块和同源基因,无论其基因结构是否完整。在 HDH 基因中,我们在旋花科物种中识别出共线性区域和同源基因的完整组装,并通过体内酶活性实验验证了其活性,这表明该基因及其功能起源于两个家族分化之前。然而,抑制 HDH 基因并未导致预期中的 山莨菪 中莨菪碱(13)的减少,而先前的研究表明,过表达 HDH 可以显著增加莨菪碱(13)的产量。这两项实验表明,HDH 基因可能对莨菪碱(13)生物合成是足够的但不是必需的,其他尚未识别的基因可能在其抑制时影响或补充其功能。
这些发现支持HS生物合成途径在茄科家族谱系分化之前的祖先起源,类似于其他植物性状的多次丧失。这一概念得到了以下两个事实的进一步支持。首先,根据HS生产物种中的共线性基因生成的基因树与物种树紧密对齐。三个HS生产物种中每个HS合成阶段的基因没有聚集成单系群,这在趋同进化的情况下会发生。其次,关键位点在三个HS生产物种中是保守的,包括 TRI 基因中五个关键位点的变异,这些位点在各种非HS生产物种的谱系中是保守的。在趋同进化的情况下,这些相似但关键的位点不太可能在缺乏该性状的近缘物种中出现在相同的基因中。功能测试支持这一观点,因为非HS生产物种中由于松散进化导致的这些关键位点的突变限制了它们的化学活性。这些位点突变的积累可能导致功能失活,加速伪基因化,并最终导致非HS生产物种中基因的丧失。我们对 StTRI 和 BaTRI 进行的功能获得和功能丧失实验通过相互突变实验为这一假设提供了有力支持。在引入HS生产物种中的突变后,我们在土豆 StTRI 中识别出酶活性的恢复。这表明某些关键位点由于非HS生产物种中的松散进化而经历了有限或终止的活动。因此,我们的系统比较和实验测试表明,在茄科祖先谱系中已经建立了单一的HS生物合成途径,随后发生了多次独立的丧失。这些丧失可能是对选择压力或在其他相关谱系中发生的遗传漂变的响应,贯穿于谱系分化和扩散过程中。生产HS的三个群体(Datureae、Hyoscyaminae 和 Mandragorinae)分布在南半球和北半球,在食草昆虫的持续选择压力下保留了这种祖先途径。
虽然我们的研究没有对所有三个HS生产物种中的HS基因活性进行检查,但我们预计这些HS基因可能具有活性。这得到了其他相同谱系的HS生产物种中有限数量HS基因的功能评估支持,并在另一项研究中报告了类似的结果。此外,在 山莨菪 上进行的四个基因的VIGS实验也提供了这些基因在HS生物合成中关键作用的额外证据。此外,我们发现,除了三个HS生产物种外,PPAR基因也在非HS生产的 Petunia 和 中国枸杞 中被识别出来。同样,HDH 基因在两个旋花科物种中被完整识别,而在其余非HS生产物种中则完全丧失。PPAR和HDH基因在这些非HS生产物种中被保留并表现出完整的功能。这些发现支持了HS生物合成在茄科家族谱系分化之前就已经存在的观点。某些谱系中途径中的基因的随机丧失导致了HS在茄科家族中的不均匀分布。然而,HS生物合成途径的共线性块和特定关键基因在某些非HS生产的茄科作物中得以保留,如土豆、番茄、烟草和辣椒,它们在全球范围内广泛种植。除了建立一种全新的HS生物合成路径,将所有相关基因转移到酵母中之外,我们的研究结果表明,将期望的HS生物合成工程化到非HS生产的茄科作物中是可行的。通过在共线性区域中对同源基因进行特定位点突变并整合缺失基因,可以在这些作物中建立HS途径,从而获得成本效益高且具有生物活性的HS化合物。
方法
植物材料、文库构建和基因组测序
此前已有报道指出,HS可以在茄科的三个谱系(Hyoscyaminae、Datureae 和 Mandragorinae)中的所有物种中检测到。为代表这些谱系,我们选择了 山莨菪、木本曼陀罗 和 茄参 作为这三个HS生产谱系的代表物种。早期研究表明,在 Lycieae 部落中的 中国枸杞 和 L. halimifolium 中存在东莨菪碱。然而,所有随后专注于检测该部落中HS的研究均未能在任何物种或样本中发现其存在。因此,我们通过额外检查确认 中国枸杞 作为非HS生产的 Lycieae 的代表物种(补充图36)。值得注意的是,另一种密切相关的同属物种 L. barbarum 的基因组序列也已报道。比较这两个物种的HS基因后,未发现任何根本性变化。因此,我们仅使用 中国枸杞 代表 Lycieae 谱系。同样,我们也排除了另一项背对背研究中的基因组序列,该研究分别使用了来自HS生产谱系Hyoscyaminae和Datureae的 A. belladonna 和 Datura stramonium 两种不同物种。该独立研究得出了类似的结论。
从 山莨菪、木本曼陀罗、茄参 和 中国枸杞 的新鲜幼叶中提取了高质量的基因组DNA,这些样本分别采自中国的门源(青海省,voucher specimens Liujq201904)、成都(四川省,Liujq201905)、果洛(青海省,Liujq202203)和兰州(甘肃省,Liujq202005)。DNA的提取采用了CTAB方法,随后使用QIAGEN DNA纯化试剂盒进行纯化。每个样本使用TruSeq Nano DNA HT样本制备试剂盒(Illumina USA)生成了短读长基因组文库,并按照制造商的说明书操作。每个样本的序列都附有独特的条形码。这些文库在Illumina NovaSeq 6000平台上进行了测序,生成150 bp的双端读长,插入片段大小约为350 bp。
对于Nanopore平台,我们选择并使用BluePippin尺寸选择系统和AMPure磁珠进行大片段的纯化。经过末端准备、测序适配子的连接和连接末端的附加,所有片段在ONT GridION X5平台上使用6个Nanopore流动池进行了测序。对于PacBio Sequel II平台,按照PacBio Sequel协议,使用BluePippin尺寸选择系统生成了SMRTbell DNA文库(~20 kb)。使用PacBio Sequel II系统构建了长读长。具体来说,我们从 山莨菪 的PromethION流动池上获得了约164 Gb的读长数据,从 中国枸杞、木本曼陀罗 和 茄参 的SMRT细胞中分别获得了约450 Gb、477 Gb 和 87 Gb 的读长数据。对于每个物种的转录组分析,使用相同个体的四种组织(叶、根、次生根和茎,每种组织为三重重复)进行RNA测序。总RNA提取使用RNeasy Plus Mini Kit(Qiagen)进行。文库构建和测序由武汉的BGI-Shenzhen公司在MGI2000平台上使用2×150 bp双端模型完成。
基因组组装与评估
使用基于k-mer的方法估算基因组大小。使用Jellyfish(v 2.3.0)和GenomeScope(v 2.0)估算基因组大小,分别使用17 bp和23 bp的k-mer大小生成k-mer深度分布。使用NextDenovo(v 1.1.1)组装程序对 山莨菪 基因组进行de novo组装。使用NextCorrect模块纠正测序错误,并基于NextGraph模块获得初步组装。使用Racon(v 1.4.10)和Pilon(v 1.23)对基因组contigs进行了进一步优化。此外,分别使用Racon和Pilon软件对Nanopore长读长和Illumina短读长进行了两次错误校正。对于 木本曼陀罗、中国枸杞 和 茄参 的组装,使用PacBio HiFi原始读长和hifiasm(v 0.12)软件进行了组装,并使用Illumina数据和Pilon进一步对基因组进行了优化。使用BUSCO(v 3.0)通过1614个来自胚胎植物(embryophyta odb10)的基因评估了基因组组装的完整性和准确性。使用Burrows-Wheeler Aligner(BWA)(v 0.7.12-r1039)和HISAT2(v 2.2.1)将Illumina DNA和RNA文库序列分别映射到组装的基因组上,以评估基因组的质量。
染色体赋值使用Hi-C技术
Hi-C(高通量染色体构象捕获)文库按照改进后的程序进行,以提高方法的有效性。具体来说,将样品的新鲜叶片暴露在含有1%甲醛的细胞核分离缓冲液中进行交联,然后用液氮将固定的组织研磨成粉末。所得的细胞核悬液被分离和纯化。随后,纯化的细胞核用100单位的HindIII消化,并通过生物素-14-dCTP标记。连接的DNA被剪切成片段,然后进行钝端修复和加A尾处理。使用生物素-链霉亲和素介导的下拉法纯化后,Hi-C文库被定量并在Illumina Hiseq平台(Illumina, San Diego, CA, USA)上进行测序。
总共生成了1,064,114,184 bp、525,935,044 bp、377,405,779 bp和273,678,964 bp的干净双端读长序列,分别来自 山莨菪、中国枸杞、木本曼陀罗 和 茄参 的文库。使用Hi-C-Pro对Hi-C原始数据进行了质量控制。使用fastp(v 0.20.0)滤除低质量序列(质量评分<20)、接头序列和短于30 bp的序列。将得到的干净双端读长序列映射到草稿组装的序列上,以获得唯一映射的双端读长序列。结果生成了268,827,577 bp、308,270,700 bp、258,535,429 bp 和167,235,469 bp 的唯一映射双端读长,分别来自 山莨菪、中国枸杞、木本曼陀罗 和 茄参。其中,80.67%、77.34%、88.13% 和 81.25% 是有效的相互作用对。结合有效的Hi-C数据,使用LACHESIS de novo组装管道生成了染色体水平的scaffolds。使用100 kb分辨率绘制了所有拟染色体的相互作用矩阵热图。
重复序列与非编码RNA注释
我们使用两种互补的方法(基于同源性和基于de novo的方法)来揭示和分类转座元件(TEs)。使用RepeatMasker(v 4.0.7)从已知重复库Repbase构建同源性重复库。使用RepeatModeler(v 1.0.11)构建基于de novo的重复库。通过比较TE蛋白数据库,使用RepeatProteinMask检测TEs。使用Tandem Repeats Finder(v 4.04)软件注释得到的整合重复库。tRNA基因的预测使用tRNAscan-SE(v 2.0)软件包。此外,rRNA、miRNA和snRNA片段的预测使用INFERNAL(v 1.0)搜索Rfam数据库进行。
基因结构与功能注释
在四个茄科基因组中使用综合方法预测蛋白编码基因,方法包括三种不同程序:(1)de novo预测、(2)基于同源性的预测和(3)基于RNA-seq的预测。de novo预测使用三个ab initio基因预测程序:Augustus(v 3.2.3)、GENSCAN(v 1.0)和GlimmerHMM(v 3.0.4)。基于同源性的预测中,六个物种(Arabidopsis thaliana、Capsicum annuum、Nicotiana tabacum、Solanum lycopersicum、Solanum pennellii 和 S. tuberosum)的蛋白质序列与重复掩码基因组对齐,利用GeMoMa(v 1.6.1)进行。我们使用两种方法生成基于RNA-seq的预测。一种方法涉及将RNA-seq数据映射到基因组并使用Tophat(v 2.1.1)和Cufflinks(v 2.2.1)组装转录本。另一种方法是使用Trinity(v 2.8.4)进行de novo组装RNA-seq数据,然后使用PASA(v 2.3.3)增强基因结构。为了确认基因集,所有预测使用EVidenceModeler(EVM v 1.1.1)合并,以生成非冗余基因集。
基因功能注释使用BLAST(v 2.3.0)搜索SwissProt和NCBI非冗余(NR)蛋白数据库。使用InterProScan(v5.36-75.0)查询包括Pfam、PRINTS、PROSITE、ProDom和SMART在内的InterPro数据库进行motif和domain注释。每个基因的Gene Ontology(GO)术语来自相应的InterPro描述。此外,将基因集映射到KEGG途径数据库以确定每个基因的最适分类。
基因家族和系统发育分析 从12个物种(即 O. sativa、V. vinifera、C. sativus、M. guttata、P. axillaris、N. attenuata、中国枸杞、山莨菪、茄参、木本曼陀罗、S. tuberosum 和 S. melongena)中获得的蛋白编码基因用于表征基因家族。使用OrthoMCL(v 2.0.9)程序识别 山莨菪、中国枸杞、木本曼陀罗、茄参 以及其余八个物种中的直系同源组。通过比较祖先和每个物种之间簇大小的差异,使用CAFÉ(v 4.2)软件识别基因家族的扩展和收缩。
为了推测系统发育位置,执行了三项程序。首先,使用MAFFT(v 7.402)对所有单拷贝基因序列进行对齐,采用默认参数。随后,使用RAxML(v 8.2.12)通过最大似然法和1000次引导复制生成12个物种的连接序列系统发育树。选择 O. sativa 作为此系统发育树的外群。最后,使用PAML(v 4.9 h)包中的MCMCtree程序基于生成的系统发育树确定发散时间。mcmctree参数设置如下:烧入量10000,样本数量100000,样本频率2。校准点从出版物和TimeTree网站获取,用于约束 V. vinifera 和 O. sativa 之间节点的年龄(160 Mya)。树图使用FigTree辅助可视化和编辑。
基因组共线性与全基因组重复分析
为了评估 山莨菪、中国枸杞、木本曼陀罗 和 茄参 基因组的进化,我们在这些茄科基因组中搜索了全基因组重复(WGD)。使用MCScanX(v 1)程序,对包含超过五个共线性基因的区域进行共线性块搜索。使用MCScan(Python版本v 1.2.7)进行补充评估。共线性基因对的Ks值计算通过使用MCScanX中的Perl脚本“add_ka_and_ks_to_collinearity.pl”实现。使用BLASTp(v 2.3.0)对来自多个基因组的所有可能染色体对的蛋白质序列锚点进行识别,E值阈值为≤1e-5。理想匹配以红色显示,其他匹配以蓝色显示,以帮助区分直系同源和旁系同源。所有点图使用WGDI(v 0.4.1)绘制。基因对的Ks值使用TBtools(v1.0987)和简单Ka/Ks计算器(MA)功能计算。
HS合成基因的识别与微共线性分析
我们使用BLAST搜索和隐马尔可夫模型(HMM)(v 3.3)在三个HS生产物种(山莨菪、木本曼陀罗 和 茄参)的基因组中挖掘同源物,采用 A. belladonna 的序列作为参考。将通过这两种方法获得的基因序列整合并手动评估,以纠正自动注释中的错误。为了探索鉴定出的HS合成基因在三个HS生产物种中的表达情况,我们在四种组织中进行了RNA测序。删除原始读长中低质量碱基百分比超过50%和未知碱基(>10%)的低质量读长,以保留干净读长。质量检查后的RNA-seq读长使用HISAT2(v 2.2.1)软件比对到四个组装的参考基因组上。使用StringTie(v 2.1.3)评估每百万映射读长的每千碱基转录本模型(TPM值)。为了简化后续分析,仅选择最终注释的基因模型进行TPM计算。转录网络分析使用WGCNA(v 1.68)进行。我们评估了基因组中的最终HS合成基因,并与先前研究中功能验证的基因进行了比较。
在本研究中,遵循Griesmann等人描述的方法,在17个所选基因组中发现了全基因组共线性块。对17对注释基因模型的蛋白质序列进行所有对所有的Blastp(E值≤1e−5),生成蛋白质相似性数据库。随后,将17个物种的基因组序列加载到JCVI实用程序库中可用的多重共线性扫描(MCSCAN,Python版本)程序中,负责创建共线性或共线性块数据库。使用LAST(v 2.32.1)进行基因对之间的比较。去除得分较低的匹配后,将从LAST输出中获得的锚点聚类为共线性块。对候选物种的每对共线性块执行的参数为“-a -e 1e-5 -s 5”。为了识别与HS合成相关的所有同源共线性块,我们采用 山莨菪、木本曼陀罗 和 茄参 的基因组作为参考,搜索并定位目标基因在共线性块中的位置,结合上下游100 kb基因组区域的侧翼基因。如果在其他对齐基因组的共线性块中识别到相应的直系同源物,则将其定义为情景1(表明共线性支持基因的存在,与全基因组基因家族直系同源物预测一致)。然而,如果在给定基因组的共线性块中未发现目标直系同源基因,则将其定义为情景2(共线性支持基因的缺失)。在完成此过程后,我们综合评估了三个基因组的发现,并更新了上述两种情景。对于未发现包含目标基因的共线性块的物种,我们手动评估蛋白质数据库并在假定区域中搜索伪基因,定义为情景3。最后,使用“synteny”功能生成微共线性图。
TRI酶活性验证
将来自 木本曼陀罗、山莨菪、茄参、中国枸杞 和 S. tuberosum 的TRI基因的编码序列,以及BaTRI(四个串联重复基因之一)、AtTRI、McTRI、LcTRI(两个串联重复基因之一)和StTRI扩增并克隆到蛋白质表达载体pET28a中。将五个TRI突变序列直接合成并克隆到pET28a中。随后,这些构建体转化到 Escherichia coli Rosetta(DE3)菌株中。经IPTG隔夜诱导后,通过离心分离裂解细菌的可溶部分。使用Ni-NTA琼脂糖根据制造商的说明纯化重组His标签TRI。通过SDS-PAGE电泳评估纯化的重组托品酮还原酶。
酶促反应按以下步骤进行。具体来说,通过分光光度法在340 nm和30°C下测量NADPH+的消耗量,以评估底物还原活性。在每个反应中,将20 µg的TRI蛋白加入到1 mL的反应缓冲液中,包含200 µM的NADPH+,5 mM的托品酮(用于确定Km值的0.01-16 mM;需要根据酶促反应结果调整每种底物的浓度范围)和0.1 M的磷酸钾(pH 6.4)。在酶促反应的初始线性阶段收集数据,以确定动力学参数。使用非线性回归的Michaelis-Menten方程计算酶促动力学常数。所有反应均进行三次,并报告平均值和标准偏差。
为了验证催化反应的最终产物,样品经过高分辨率质谱分析。样品通过UPLC-MS分析,用ACQUITY UPLC BEH HILIC柱分离,使用电喷雾电离在正离子模式下进行MS分析。
LS酶活性检测
由于UGT1生成的苯基乳糖苷产量有限,难以分离出足够数量供LS酶活性检测。因此,我们选择了植物中的瞬时表达方法来研究来自两个生产HS的物种(即山莨菪的AtLS和木本曼陀罗的BaLS)的LS基因的预期功能。作为对照,我们使用了来自同一谱系的生产HS的A. belladonna的AbLS,其功能已被实验证实。此外,我们还包括了来自该物种的AbUGT1用于后续的化学反应。将YFP(对照)、AbUGT1、AbLS、AtLS和BaLS的全长编码序列(CDS)插入pEAQ-HT载体中,使用AgeI和XhoI限制性内切酶。所有使用的引物列表详见补充表30。
随后,这些构建体分别转化到Agrobacterium菌株GV3101中。将改造后的Agrobacterium分别培养后,将细菌悬浮在含有10 mM MgCl2和150 mM乙酰丁香酮的缓冲液中,并将菌液的OD600调整至0.6。溶液在室温下孵育3小时。将菌液充分混合后注入烟草叶片中。96小时后,将烟草叶片渗入含有1 mM托品酮(CAS:120-29-6)和1 mM苯基乳糖苷(CAS:7326-19-4)的底物溶液中。渗透后的烟草叶片在24小时后收获并冻干以进行代谢物分析。每个实验组包括六个生物学重复。使用Thermo Scientific UltiMate 3000 UPLC系统与Thermo Scientific Q Exactive Orbitrap LC-MS仪器和Hypersil GOLD C18柱(100 mm×2.1 mm,1.9 µm)在40°C的温度下检查littorine的存在。
PPAR酶活性检测
分别使用ApexHF HS DNA聚合酶(Accurate Biology,武汉,中国)通过PCR扩增PPARs的编码序列。随后,扩增的序列被插入E. coli表达载体pET28a中。用于克隆PPARs的所有引物列表详见补充表31。根据制造商的说明,使用Ni-NTA琼脂糖(QIAGEN,德国)纯化重组的His标签PPARs。纯化后,PPARs(20 µg)酶在冷透析液(50 mM磷酸钾,pH 8.0)中透析,然后在30°C下与3 mM苯基丙酮酸(CAS:156-06-9)和2 mM NADPH(CAS:2646-71-1)孵育2小时。煮沸的酶样品用作阴性对照。最后,通过加入等体积的甲醇停止反应。使用Thermo Scientific UltiMate 3000 UPLC系统和Thermo Scientific Q Exactive Orbitrap LC-MS仪器检测催化产物苯基乳酸。
CYP80F1和HDH酶活性检测
由于缺乏标准化的HDH酶活性检测底物,我们利用植物中的瞬时表达方法来检测来自三个生产HS的物种(山莨菪、木本曼陀罗和茄参)的CYP80F1和HDH基因的预期功能。此外,我们验证了来自不同非生产HS物种的共线性CYP80F1和HDH基因的功能。将CYP80F1和HDH的CDS插入pEAQ-HT载体中,使用AgeI和XhoI限制性内切酶或OK Clon DNA连接技术。我们使用了来自A. belladonna的AbCYP80F1和AbHDH基因作为对照。这些构建体随后分别转化到Agrobacterium GV3101中。CYP80F1和HDH基因在烟草中的感染实验按照LS基因的步骤进行。注射菌液后,将烟草叶片浸入含有NADPH和littorine底物的溶液中。随后,将浸入底物的烟草叶片在24小时后收获并冻干以进行代谢物分析。使用Thermo Scientific UltiMate 3000 UPLC系统和Thermo Scientific Q Exactive Orbitrap LC-MS仪器通过Hypersil GOLD C18柱检测产物。
H6H酶活性验证
由于已确认山莨菪和木本曼陀罗中H6H基因的酶活性,我们专注于通过体外酶促反应评估中国枸杞和茄参中的H6H酶活性。将茄参和中国枸杞的H6H基因的CDS分别克隆到蛋白质表达载体pET28a中。所有使用的引物详见补充表33。随后,这些构建体被转化到E. coli菌株BL21 star(DE3)中。
所有纯化程序在0-4°C下进行,使用Ni-NTA琼脂糖根据制造商的说明进行纯化。纯化后的H6Hs酶在冷透析液(50 mM Tris-HCl,pH 7.4)中透析。通过测量从各种底物形成的产物来评估H6Hs的酶活性。完整的催化反应混合物由50 mM Tris-HCI缓冲液(pH 7.4),0.4 mM FeSO4,4 mM抗坏血酸钠,1 mM 2-氧代戊二酸,0.2 mM l-东莨菪碱氢溴酸盐或0.2 mM 东莨菪碱,2 mg/mL过氧化氢酶和酶组成。在30°C下孵育催化反应系统2小时后,加入NaHCO3将混合物的pH提高至9.0以终止反应。混合物被三次等量的乙酸乙酯提取,提取物在55°C下蒸发。最后,将残留物溶解在0.5 ml甲醇中,并储存在4°C以供后续使用。通过Agilent 1260 infinity II高效液相色谱(HPLC)使用ZORBAX Eclipse XDB-C18柱进行生物碱分析。
TRI、LS、HDH和H6H基因在山莨菪中的病毒诱导基因沉默(VIGS)
VIGS是一种强大的中等高通量植物基因功能表征方法。它被认为是一种在茄科植物中特别有效的方法,并已成功用于沉默与生物碱合成相关的根表达基因。在本研究中,我们手动去除了从高海拔野外采集的山莨菪种子的种皮,并将其放置在湿润的滤纸上进行发芽。一旦胚根出现,幼苗被移植到营养土中,并在24°C下的16小时光照/8小时黑暗周期中生长。大约一个月后,当第二片真叶出现时,进行VIGS测试。
VIGS使用双组分TRV系统进行。通过连接体克隆将目标基因构建体插入TRV2-LIC载体中。使用补充表34中描述的引物对从幼苗的gDNA扩增山莨菪 PDS、TRI、LS、HDH和H6H的PCR片段。将这些片段直接连接到pTRV2载体中,生成了命名为pTRV2-AtPDS、pTRV2-AtTRI、pTRV2-AtLS、pTRV2-AtHDH和pTRV2-AtH6H的构建体。通过PCR验证和DNA序列分析验证重组克隆。pTRV1、pTRV2、pTRV2-AtPDS、pTRV2-AtLS、pTRV2-AtTRI、pTRV2-AtHDH和pTRV2-AtH6H的构建体分别转化到Agrobacterium tumefaciens菌株GV3101中。48小时后,选择单菌落于1 ml的YEP培养基中,并补充50 mg/L利福平(Rif)、50 mg/L卡那霉素(Kan)和10 mg/L硫酸庆大霉素(Gen)在28°C下进行隔夜激活。经过PCR
检测后,将细菌接种到100 mL(Kan+Rif+Gen)YEP液体培养基中,浓度为2%,并在28°C下培养过夜。
第二天,将活化的细菌以4500 rpm/min的速度离心10分钟。弃去上清液,并将细菌重悬于缓冲液中,OD600=1.0。悬浮液在室温下静置3至5小时。在注射前,将pTRV1与所有pTRV2重组载体的A. tumefaciens菌株GV3101以1:1的比例混合。最后,通过使用两种方法将Agrobacterium培养物渗入山莨菪幼苗的子叶中来进行沉默实验。第一种方法是叶片注射,使用消毒过的针头抽取菌液并注射到真叶的背侧静脉中。第二种方法是根灌,使用消毒的移液器抽取5 mL菌液,然后将其插入根部附近的土壤中。每7天重复渗透,且每种方法进行三次。渗透后,将植物在黑暗中培养48小时,然后转移到生长室,在24°C下保持16小时光周期。28天后,获得感染植物的根部以进行基因表达分析。使用六个独立的表达下调线进行后续的生物碱测量。在感染植物后45天收集用于生物碱提取的组织样本。
使用适量的山莨菪感染植物组织,在液氮中研磨。使用植物RNA提取试剂盒(BIOFIT,中国)提取总RNA。使用Hifair® III 1st Strand cDNA合成SuperMix for qPCR合成第一链cDNA。PGK基因用作评估每个目标基因表达的内部参考。所有引物序列可在补充表34中找到。对于qRT-PCR反应,使用Hieff UNICON® Universal Blue qPCR SYBR Green Master Mix。
感染植物中生物碱的提取和检测按照方法进行。首先,从每条线上分离代谢物。使用LC-MS/MS系统检测和定量托品生物碱,系统配有Thermo Hypersil Gold分析柱(100 mm×2.1 mm,1.9 µm),在40°C下运行。定量分析通过在正离子模式下使用多反应监测的电喷雾电离进行。与每个样品集一起评估一系列校准标准和空白。
代谢物提取和检测
代谢物提取按照以下方法进行。最初,将植物样品冻干并细粉化为粉末。从这些冷冻粉末中使用每25 mg样品粉末1 mL的含20%甲醇和0.1%甲酸的提取缓冲液提取代谢物。以13,000 × g离心5分钟后,收集所得上清液以进行UPLC-MS分析。使用UPLC-MS检测托品、littorine、苯基乳酸和东莨菪碱。仪器参数如下:源电压为3.0 kV,毛细管温度为350°C,S-lens RF水平为50,辅助气体加热器温度为350°C,流速为0.5 mL/min,流动相由含0.1%甲酸的水(泵A)和乙腈(泵B)组成。
对于东莨菪碱和莨菪碱的检测,使用与ZORBAX Eclipse XDB-C18柱配合的HPLC系统。流动相含有0.02%的甲酸。littorine、苯基乳酸、东莨菪碱、东莨菪碱和莨菪碱的洗脱程序详见补充表35和36。对于中国枸杞中生物碱的检测,最初将用于测量的不同组织研磨成粗粉并在60°C下干燥8小时。为了从该粉末中提取代谢物,使用包含0.2 g NaCl2、8.6 ml乙醇、1 ml水和0.4 ml甲酸的提取缓冲液。在超声处理5分钟后,收集所得上清液用于通过Wondasil C18柱进行的HPLC-MS分析。
HS合成基因关键位点的祖先状态推断
使用FastML软件对17个茄科物种的TRI比对的氨基酸序列进行最大似然祖先状态推断。推断中使用了在序列模拟中使用的相同氨基酸频率、alpha和氨基酸替换模型。保留了在最大似然框架内具有最高概率的状态作为推断的祖先状态。
这篇关于茄科四个参考基因组-文献精读41的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!