本文主要是介绍「BioNano系列」光学图谱混合组装应该怎么做?,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
评估从头组装结果
Bionano从头组装出光学图谱CMAP可以和参考序列的CMAP进行比对,通过Access上可视化检查参考基因组的组装质量,比较两者间的不同。
这里所用的CMAP图谱来自于一篇发表在NC的拟南芥的基因组文章(原本计划用他们的bnx文件介绍从头组装,但是通讯作者根本不搭理我),
光学图谱的下载方式为:
wget https://submit.ncbi.nlm.nih.gov/ft/byid/w4jcevedkbs-mac-74_bng_contigs2017.cmap
我通过Canu以原始错误率0.5纠错后直接以纠错后错误率0.144进行组装, 得到的物理图谱, 可通过百度网盘(链接:https://pan.baidu.com/s/1PGYvCE0Ku65vwNQ3cEscKA 提取码:88us )进行下载。
分析代码如下:
#模拟酶切
perl /opt/biosoft/Solve3.3_10252018/Pipeline/10252018/fa2cmap_multi_color.pl -i R05C0144.fa -e BspQI 1
# 两个图谱比较
python /opt/biosoft/Solve3.3_10252018/Pipeline/10252018/runCharacterize.py \-t /opt/biosoft/Solve3.3_10252018/RefAligner/7915.7989rel/RefAligner \-q kbs-mac-74_bng_contigs2017.cmap -r R05C0144_BSPQI_0kb_0labels.cmap \-p /opt/biosoft/Solve3.3_10252018/Pipeline/10252018 \-a /opt/biosoft/Solve3.3_10252018/RefAligner/7915.7989rel/optArguments_nonhaplotype_noES_irys.xml \-n 10
运行之后会在当前目录下生成一个"alignref"文件夹, 将其中的"q.cmap","r.cmap",".xmap"下载到本地,上传到access中进行可视化
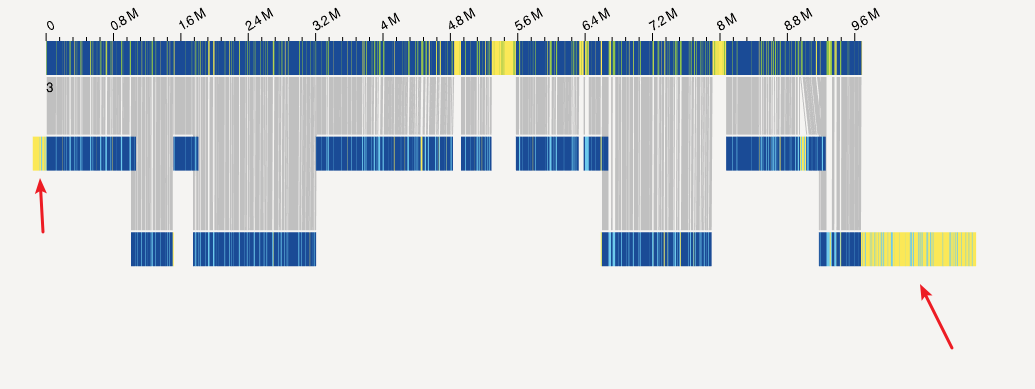
上图中,箭头指示的部分可能就是光学图谱能用于锚定其他contig的部分,这就是下一节光学图谱辅助组装的原理。
光学图谱辅助组装
NGM(Next-Generation Mapping) Scaffold 流程:
- 为序列数据产生 in silico 图谱
- 将序列和Bionano基因组图谱进行比较,找到两者之间的冲突并尝试解决
- 将不冲突的图谱合并成 hybrid scafold
- 在序列图谱和hybrid scaffold之间形成联配
- 得到scaffold的AGP和FASTA文件
整个流程和Bionano Access完美整合,为使用者提供了方便的操作界面,用于对scafflod结果进行可视化。流程的脚本在"/opt/biosoft/Solve3.3_10252018/HybridScaffold/10252018"
单酶系统
流程控制脚本为: Solve3.3_版本日期HybridScaffold/版本日期/hybridScaffold.pl, 他接受输入文件,输出运行过程中的信息,产生输出文件,最后得到结果描述。
有四个必须文件: FASTA格式组装结果,CMAP格式的Bionano 基因组图谱组装,XML格式的配置文件, RefAligner.
perl hybridScaffold.pl -n FASTA格式序列 (必须)-b BIonano CMAP文件 (必须)-c Merge 的XML配置文件 (必须)-r RefAligner运行工具路径 (必须)-o 输出文件夹 (必须)-B conflict filter level genome maps; 1,2 or3, 决定如何处理冲突,1表示不过滤,2表示在冲突处分割contig,3表示删除冲突的contig,没有-M时一定要加入-N conflict filter level for sequences; 1,2 or 3, 决定如何处理冲突,1表示不过滤,2表示在冲突处分割contig,3表示删除冲突的contig,没有-M时一定要加入-f 是否覆盖之前的输出-x 分别进行hybrid scaffold 和 genome map的相互比对-y 为输入的genome maps生成嵌合质量分-M 输入手工解决过冲突的文件-m: 如果使用了-x或-y参数,则需要输入Bionano molecules的BNX文件-p 从头组装流程的文件路径,如果使用了-x或, -y 选项,就需要加入这一项-q 从头组装流程的XML配置文件,如果使用了-x或, -y 选项,就需要加入这一项-e 从头组装时的噪音参数, .errbin或err文件-v 输出流程版本信息
明确一点: -c 要求的XML文件真的不是无脑用,需要修改其中fasta2cmap的enzyme部分
实际运行案例:
cp /opt/biosoft/Solve3.3_10252018/HybridScaffold/10252018/hybridScaffold_config.xml .
# 用vim修改hybridScaffold_config.xml中的enzyme
perl /opt/biosoft/Solve3.3_10252018/HybridScaffold/10252018/hybridScaffold.pl \-n R05C0144.fa \-b kbs-mac-74_bng_contigs2017.cmap \-c hybridScaffold_config.xml \-r /opt/biosoft/Solve3.3_10252018/RefAligner/7915.7989rel/RefAligner \-o R05C0144 \-B 2 -N 2 \-f
运行过程中会输出scaffold N50等一些参数。N50仅仅提升了1.1M,估计是作者bionano数据不够多。
组装的FASTA在"R05C0144/agp_fasta"文件下,而"R05C0144/hybridScaffold_archive.tar.gz"可以上传到Access查看组装效果, 下图就是一个典型的混合组装

当然具体分为哪几步,以及每一步调用的脚本如下所示:
第一步: 将FASTA转成CMAP格式,
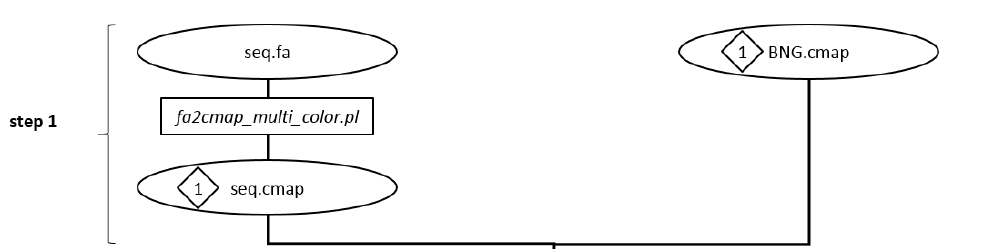
用到一个perl脚本, fa2cmap_multi_color.pl, 通过对基因组序列进行模式搜索寻找可能的酶切位点,默认输出在"fa2cmap"文件夹下
第二步: 识别并解决冲突。
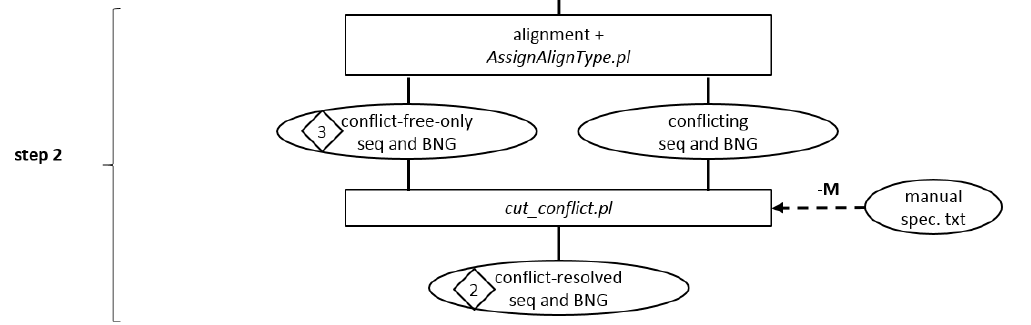
冲突可能来自于真实的等位基因,或者时组装错误,最终的结果就是在联配中出现过多无法比对上的标记(labels). Hybrid Scaffold流程会先用RefAligner将第一步得到的cmp去跟Bionano基因组图谱比,然后用AssignAlignType.pl识别冲突交界处。输入文件为RefAligner运行后得到的XMAP和CMAP文件,以及原始序列和原始Bionano基因组图谱。统计每个联配中比对和未必对标记数,根据XML配置文件中"assignAlignType.max_overhang" 参数设置最大可以容忍的无法联配的标记数。最后会输出"assginAlignType.xmap"(列出冲突位置),以及"assignAlignType_r.cmap"(无冲突序列), "assignAlignType_q.cmap"(无冲突图谱)。更重要的是"conflicts.txt",记录着每个可能的位置。
之后流程用cut_conflicts.pl解决不一致的位置, 输出"conflicts_cut_status.txt", 可以手工编辑,有监督的进行处理。
第三步: 合并两者的组装结果,形成Hybrid scaffold

这一步用MergeNGS_BN.pl脚本完成,它会调用RefAligner进行迭代两两配对合并,输入文件是下面的其中一个
- 原始输入
- 冲突解决后的组装(
cut_conflicts.pl输出结果) - 没有冲突的组装(
AssignAlignType.pl的结果)
每一种输入都是一种选项,我们可以尝试不同的输入,最后进行比较。
第四步: 将序列图图谱和基因组图谱比对到hybrid scaffold

第五步: 生成hybrid scaffold表征的AGP和FASTA文件

一些注意事项:
- Bionano很难处理Hi-C数据引起的基因组中朝向/排序的错误。所以先Bionano混合组装,然后才是Hi-C
- 覆盖度: 至少50X,NLRS随着覆盖度提高并不会有明显增强图谱连续性,DLS(例如DLE0-1) 100X以上的覆盖度能够明显提高某些植物和东西的图谱连续性。
- 当前的Hybrid Scaffold 流程无法很好处理单倍体信息,所以上一步的从头组装一定要是nonhaplotype.
这篇关于「BioNano系列」光学图谱混合组装应该怎么做?的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!








