本文主要是介绍AUCell和AddModuleScore函数进行基因集评分,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
AUCell 和AddModuleScore 分析是两种主流的用于单细胞RNA测序数据的基因集活性分析的方法。这些基因集可以来自文献、数据库或者根据具体研究问题进行自行定义。
AUCell分析原理:
1、AUCell分析可以将细胞中的所有基因按表达量进行排序,生成一个基因排名列表,表达量越高的基因排名越靠前。
2、接下来对每个基因集中的基因找到它们在每个细胞的基因排名列表中的位置,这些位置则反映了基因集内基因在特定细胞群中的表达情况。
3、计算基因集在每个细胞中的活性评分。基于基因集内基因的排名,通过计算这些基因排名的累计面积(AUC,Area Under the Curve)来评估基因集的活性。AUC值越大,表明该基因集在该细胞中的表达越活跃。
AddModuleScore分析原理:
1、计算每个基因集中的基因在每个细胞中的平均表达值
2、选择与目标基因集大小相似的背景基因集,通过目标基因集的平均表达值减去背景基因集的平均表达值,得到基因模块评分(这个评分代表)。这个背景基因集是有函数内部把表达矩阵自行切割若干份之后随机抽取每一份中的基因作为背景基因集。
应用场景
-
细胞类型鉴定:识别不同细胞类型或细胞亚群的特征基因集活性。
-
功能状态分析:分析细胞的功能状态,例如细胞周期、免疫反应等。
-
生物学过程探索:探索特定生物学过程中基因集的表达活性。
AUCell分析步骤:
1.读取基因集数据(采用了Autophagy基因数据)
rm(list = ls())
autophagy_genes <- read.xlsx("Autophagy.xlsx",colNames = T)
g <- autophagy_genes$Symbol
head(autophagy_genes)
#GeneId Name Symbol
#1 55626 autophagy/beclin-1 regulator 1 AMBRA1
#2 8542 apolipoprotein L, 1 APOL1
#3 405 aryl hydrocarbon receptor nuclear translocator ARNT
#4 410 arylsulfatase A ARSA
#5 411 arylsulfatase B ARSB
#6 468 activating transcription factor 4 (tax-responsive enhancer element B67) ATF4
2.读取R包和需要分析的数据(采用了单细胞pbmc数据)
library(Seurat)
library(tidyverse)
library(openxlsx)
load("step1.final.Rdata") #pbmc数据
sce <- step1.final
#check一下
DimPlot(sce,group.by = "celltype",label = T)+ NoLegend() +ggsci::scale_color_d3()
3、AUCell分析
library(GSEABase)
geneSets <- GeneSet(g, setName="autophagy")
geneSets
#BiocManager::install("AUCell")
library(AUCell)
exprMatrix = sce@assays$RNA@data
rownames(exprMatrix) = Features(sce)
colnames(exprMatrix) = Cells(sce)
#对矩阵中的每个细胞里,给基因进行排序(可见下图1)
cells_rankings <- AUCell_buildRankings(exprMatrix,, plotStats=TRUE)
#把每个细胞中的基因表达量从高到底排列并计算数量
#Quantiles for the number of genes detected by cell:
#(Non-detected genes are shuffled at the end of the ranking. Keep it in mind when choosing #the threshold for calculating the AUC).
# min 1% 5% 10% 50% 100%
# 491.00 633.15 806.00 897.30 1323.00 5418.00
cells_AUC <- AUCell_calcAUC(geneSets, cells_rankings,aucMaxRank = nrow(cells_rankings)*0.05)
#为了计算AUC,默认情况下只使用排名中前5%的基因(即检查基因集中的基因是否在前5%之内)。
#这样可以在更大的数据集上更快地执行,并减少排名底部噪音的影响(例如,许多基因可能表达量为0)。要考虑的百分比可以通过参数 aucMaxRank 参数进行修改。可以通过AUCell_buildRankings提供的直方图进行辅助判断。
#Genes in the gene sets NOT available in the dataset:
# autophagy: 26 (12% of 222)
set.seed(123)
#见下图2
cells_assignment <- AUCell_exploreThresholds(cells_AUC, plotHist=TRUE, assign=TRUE)
auc_thr = cells_assignment$HMMR$aucThr$selected
auc_thr
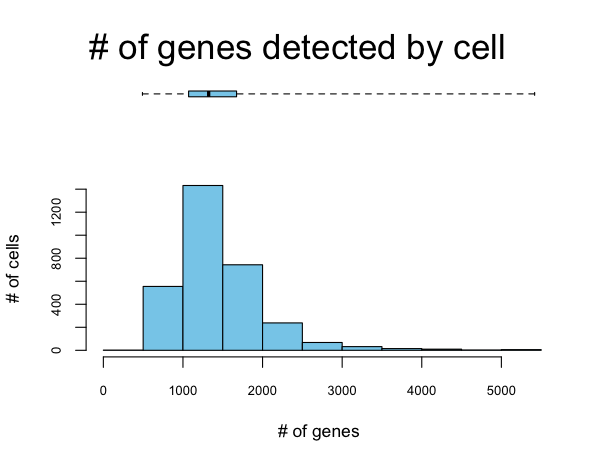
不同细胞中基因表达情况呈偏态分布
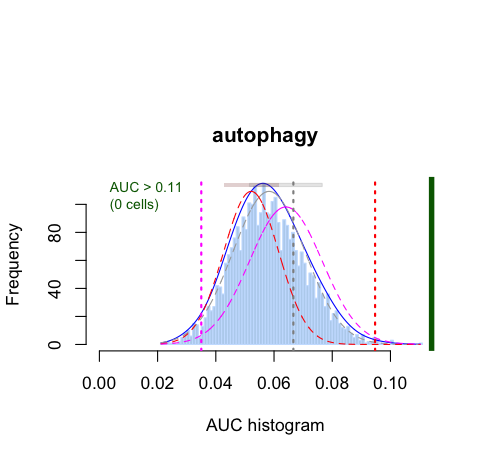
这里重点提一下如何看这个AUC histogram,我这边采用autophagy基因集在pbmc数据集中进行分析发现,AUC大于0.11的细胞为活性细胞,但是pbmc中没有细胞符合要求~ 这也提示了我们在做AUCell分析前,需要仔细考虑纳入分析的基因集和单细胞数据是否”合适“。
接下来我以AUCell github上的资料为例子,这些AUC柱状图是对“自定义/活性基因集的细胞”的直观展示。开发者采用了神经细胞的数据集进行分析和展示。
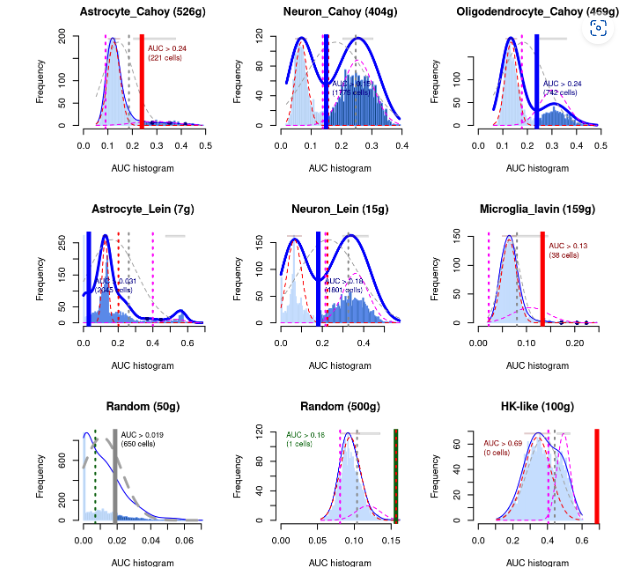
1) 图片的标题是指不同的细胞亚群和基因数量,比如Astrocyte_Cahoy (526g),星形胶纸细胞,代表这群细胞在研究者纳入分析的数据集中存在的基因数量为526个。其中Random(50g), 代表研究者随机提取细胞和基因。
2) X轴代表了AUC值的大小,Y轴代表了不同AUC值下的细胞数量,图形里边的AUC值代表了阈值,AUC阈值下边的具体数值代表了达到要求的“活性”细胞数量。
3) 理想的情况图形呈双峰分布,数据集中大多数细胞是呈现较低的AUC值,而少部分细胞则呈现较高的AUC值。比如Oligodendrocyte_Cahoy分析结果。比较类似的结果是Neuron和Microglia_lavin,虽然它们的基因集细胞数量很少或者符合要求的细胞数很少,但结果仍旧呈现出了双峰形态,或许侧面说明了他们的基因集或者筛除的细胞能够明显的表征某些特性。
4) 如果基因集是数据集中的高比例细胞的标记(比如 Neuron结果),那么分布图形可能类似于管家基因的分析结果(HK-like)。
5) 基因集的大小会影响结果。更大的基因集(100-2k)可会使结果更稳定,更易评估,随着基因数目的减少,AUC = 0的细胞数目可能增加。当然如果选定的基因集非常给力,那么也可能呈现较好的结果 (即Neuron_Lein结果)。
4、图形可视化
#由于我这里使用的autophagy基因并不能很好的区分高低活性细胞,因此我更换了数据集
#数据集采用了GSE162025鼻咽癌数据集中的上皮细胞
#基因集采用了"SCNM1","CDC42SE1","ZNF687" (随意指定)
Idents(sce) <- sce$celltype
sce$auc_score = as.numeric(getAUC(cells_AUC))
sce$auc_group = ifelse(sce$auc_score>auc_thr,"high_A","low_A") #自行修改dat<- data.frame(sce@meta.data, sce@reductions$umap@cell.embeddings,seurat_annotation = sce@active.ident)
class_avg <- dat %>%group_by(seurat_annotation) %>% #按照seurat_annotation列(即细胞的分类)对数据进行分组。summarise(umap_1 = median(umap_1),umap_2 = median(umap_2) #对每个分组计算UMAP坐标的中位数 画label)library(ggpubr)
ggplot(dat, aes(umap_1, umap_2)) +geom_point(aes(colour = auc_score)) +viridis::scale_color_viridis(option="A") +ggrepel::geom_label_repel(aes(label = seurat_annotation),data = class_avg,label.size = 0,segment.color = NA)+theme_bw()
ggsave(filename="Aucell.pdf",width = 12,height = 7)# 高低分组
ggplot(dat,aes(x = umap_1,y = umap_2))+geom_point(aes(color = auc_group),size = 0.5)+theme_classic()
ggsave(filename="Aucell2.pdf",width = 8,height = 7)
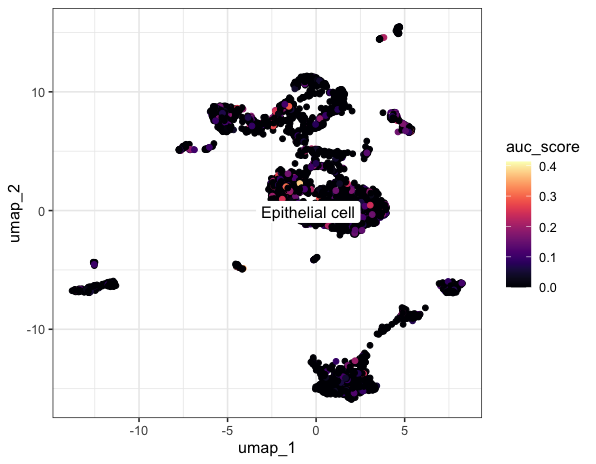
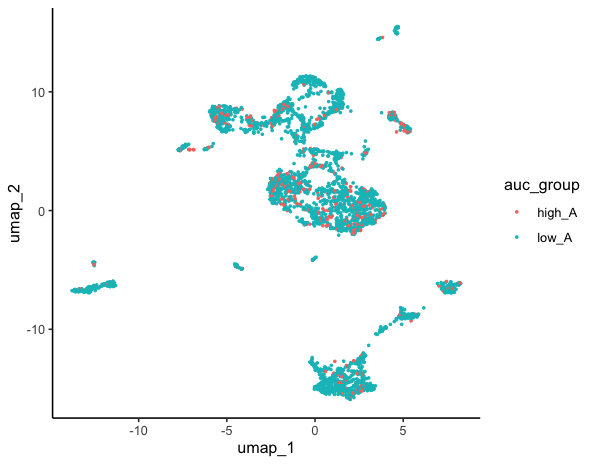
然后接下来可以根据基因集的高低分组进行更多的个性化分析啦~
AddModuleScore分析步骤:
1、读取数据和基因集
rm(list = ls())
library(Seurat)
g <- c("SCNM1","CDC42SE1","ZNF687")
load("sce_epi.Rdata") #数据集采用了GSE162025鼻咽癌数据集中的上皮细胞
#DimPlot(sce,group.by = "celltype",label = T)+ NoLegend() +ggsci::scale_color_d3()
2、AddModuleScore分析
#AddmoduleScore函数是seraut包自带的很方便
sce = AddModuleScore(object = sce,features = g)
colnames(sce@meta.data)
p =FeaturePlot(sce,'Cluster1') #默认名称cluster1
p dat<- data.frame(sce@meta.data, sce@reductions$umap@cell.embeddings,seurat_annotation = sce@active.ident)
class_avg <- dat %>%group_by(seurat_annotation) %>% #按照seurat_annotation列(即细胞的分类)对数据进行分组。summarise(umap_1 = median(umap_1),umap_2 = median(umap_2) #对每个分组计算UMAP坐标的中位数 画label)library(ggpubr)
ggplot(dat, aes(umap_1, umap_2)) +geom_point(aes(colour = Cluster1)) + #修改这里的colourviridis::scale_color_viridis(option="A") +ggrepel::geom_label_repel(aes(label = seurat_annotation),data = class_avg,label.size = 0,segment.color = NA)+theme_bw()
ggsave(filename="Aucell-addmodulescore.pdf",width = 12,height = 7)
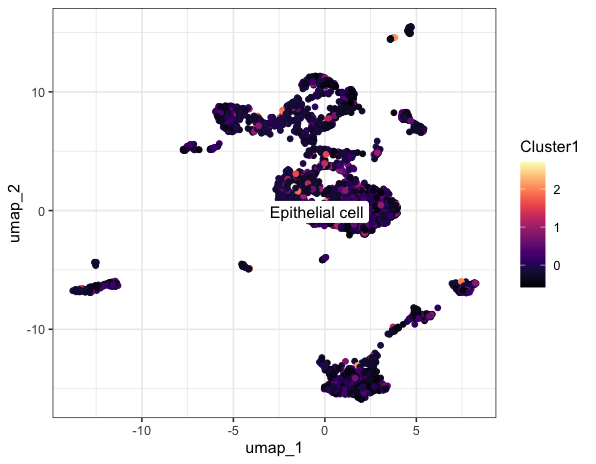
获得cluster评分之后就可以按照中位值或者其他的值进行分组,从而进行后续的个性化分析啦~
注:若对内容有疑惑或者有发现明确错误的朋友,请联系后台(希望多多交流)。更多内容可关注公众号:生信方舟
- END -
这篇关于AUCell和AddModuleScore函数进行基因集评分的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!


