本文主要是介绍R调用Taxonkit展示系统发育信息,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
Introduction
TaxonKit是一个用于处理生物分类学数据的命令行工具。
它的主要功能是处理NCBI的生物分类学数据,包括对分类单元(如物种、属、科等)的查找、分类单元的上下位关系查询、分类单元名称的标准化等。
为了方便R社区用户(自己)使用和流程整合,我把Taxonkit工具整合进了R包pctax,也开发了一些配套的系统发育分析和可视化方法。
R调用Taxonkit
准备工作
- 安装
pctax
pctax稳定版本可在CRAN上获得:
install.packages("pctax")
或者你可以通过以下方式从GitHub安装pctax的开发版本:
# install.packages("devtools")
devtools::install_github("Asa12138/pctax")
- 安装taxonkit:
library(pctax)
pctax::install_taxonkit(make_sure = TRUE)#成功后taxonkit会安装在下面这个目录👇
tools::R_user_dir("pctax")
- 下载NCBI Taxonomy数据文件:
pctax::download_taxonkit_dataset(make_sure = TRUE)#成功后Taxonomy数据文件会在下面这个目录👇
file.path(Sys.getenv("HOME"), ".taxonkit")
该函数会下载官网最新版本的Taxonomy数据库,如果需要制定版本的数据库,可以自己在官网下载:https://ftp.ncbi.nih.gov/pub/taxonomy/,然后指定位置:
pctax::download_taxonkit_dataset(make_sure = TRUE,taxdump_tar_gz = "~/Downloads/taxdump.tar.gz")
使用
# 下列命令不报错说明可以正常使用
check_taxonkit(print = FALSE)
主要功能与taxonkit一致:
| 函数 | 功能 |
|---|---|
taxonkit_list | 列出指定TaxId下所有子单元的的TaxID |
taxonkit_lineage | 根据TaxID获取完整谱系(lineage) |
taxonkit_reformat | 将完整谱系转化为“界门纲目科属种株”的自定义格式 |
taxonkit_name2taxid | 将分类单元名称转化为TaxID |
taxonkit_filter | 按分类学水平范围过滤TaxIDs |
taxonkit_lca | 计算最低公共祖先(LCA) |
并且help(taxonkit_*)可查看详细使用说明。
# 列出[genus] Homo下的所有子单元
taxonkit_list(ids = c(9605), indent = "-", show_name = TRUE, show_rank = TRUE)
## [1] "9605 [genus] Homo"
## [2] "-9606 [species] Homo sapiens"
## [3] "--63221 [subspecies] Homo sapiens neanderthalensis"
## [4] "--741158 [subspecies] Homo sapiens subsp. 'Denisova'"
## [5] "-1425170 [species] Homo heidelbergensis"
## [6] "-2665952 [no rank] environmental samples"
## [7] "--2665953 [species] Homo sapiens environmental sample"
## [8] "-2813598 [no rank] unclassified Homo"
## [9] "--2813599 [species] Homo sp."
## [10] ""
taxonkit_lineage, taxonkit_reformat, taxonkit_name2taxid, taxonkit_filter 与 taxonkit_lca 默认从文件中读取数据,也可通过指定text = TRUE从字符串输入读取输入数据:
# 查询9606和63221的完整谱系
taxonkit_lineage("9606\n63221", show_name = TRUE, show_rank = TRUE, text = TRUE)%>%pcutils::strsplit2(split = "\t",colnames = c("taxid","lineage","name","level"))
## taxid
## 1 9606
## 2 63221
## lineage
## 1 cellular organisms;Eukaryota;Opisthokonta;Metazoa;Eumetazoa;Bilateria;Deuterostomia;Chordata;Craniata;Vertebrata;Gnathostomata;Teleostomi;Euteleostomi;Sarcopterygii;Dipnotetrapodomorpha;Tetrapoda;Amniota;Mammalia;Theria;Eutheria;Boreoeutheria;Euarchontoglires;Primates;Haplorrhini;Simiiformes;Catarrhini;Hominoidea;Hominidae;Homininae;Homo;Homo sapiens
## 2 cellular organisms;Eukaryota;Opisthokonta;Metazoa;Eumetazoa;Bilateria;Deuterostomia;Chordata;Craniata;Vertebrata;Gnathostomata;Teleostomi;Euteleostomi;Sarcopterygii;Dipnotetrapodomorpha;Tetrapoda;Amniota;Mammalia;Theria;Eutheria;Boreoeutheria;Euarchontoglires;Primates;Haplorrhini;Simiiformes;Catarrhini;Hominoidea;Hominidae;Homininae;Homo;Homo sapiens;Homo sapiens neanderthalensis
## name level
## 1 Homo sapiens species
## 2 Homo sapiens neanderthalensis subspecies
从文件中读取数据:
names <- system.file("extdata/name.txt", package = "pctax")
taxonkit_name2taxid(names, name_field = 1, sci_name = FALSE, show_rank = FALSE)%>%pcutils::strsplit2(split = "\t",colnames = c("name","taxid"))
## name taxid
## 1 Homo sapiens 9606
## 2 Akkermansia muciniphila ATCC BAA-835 349741
## 3 Akkermansia muciniphila 239935
## 4 Mouse Intracisternal A-particle 11932
## 5 Wei Shen
## 6 uncultured murine large bowel bacterium BAC 54B 314101
## 7 Croceibacter phage P2559Y 1327037
系统发育树
如果是做16S测序的话,在分析过程中就会得到一个带距离的系统发育树。宏基因组分析如果组装MAG后用GTDB-Tk比对数据库后也可以获得有距离的系统发育树。
但有时候我们想要从物种名或taxid获取整齐的谱系信息,用来一个构建系统发育树(层级树,没有真实的距离,只展示包含关系)。这是一个常见的需求,很多文章都会画一个这样的树图来展示自己的数据。
可以实现这个需求的工具有一些:
- iPhylo:https://iphylo.net/,免费,快速,支持NCBI taxonomy和一些化学物质分类树,赞
- R包
taxtree,很慢 - PhyloT:https://phylot.biobyte.de/,收费
当然可以使用pctax包快速完成,对于分析流程都在R里做的人来说非常方便:
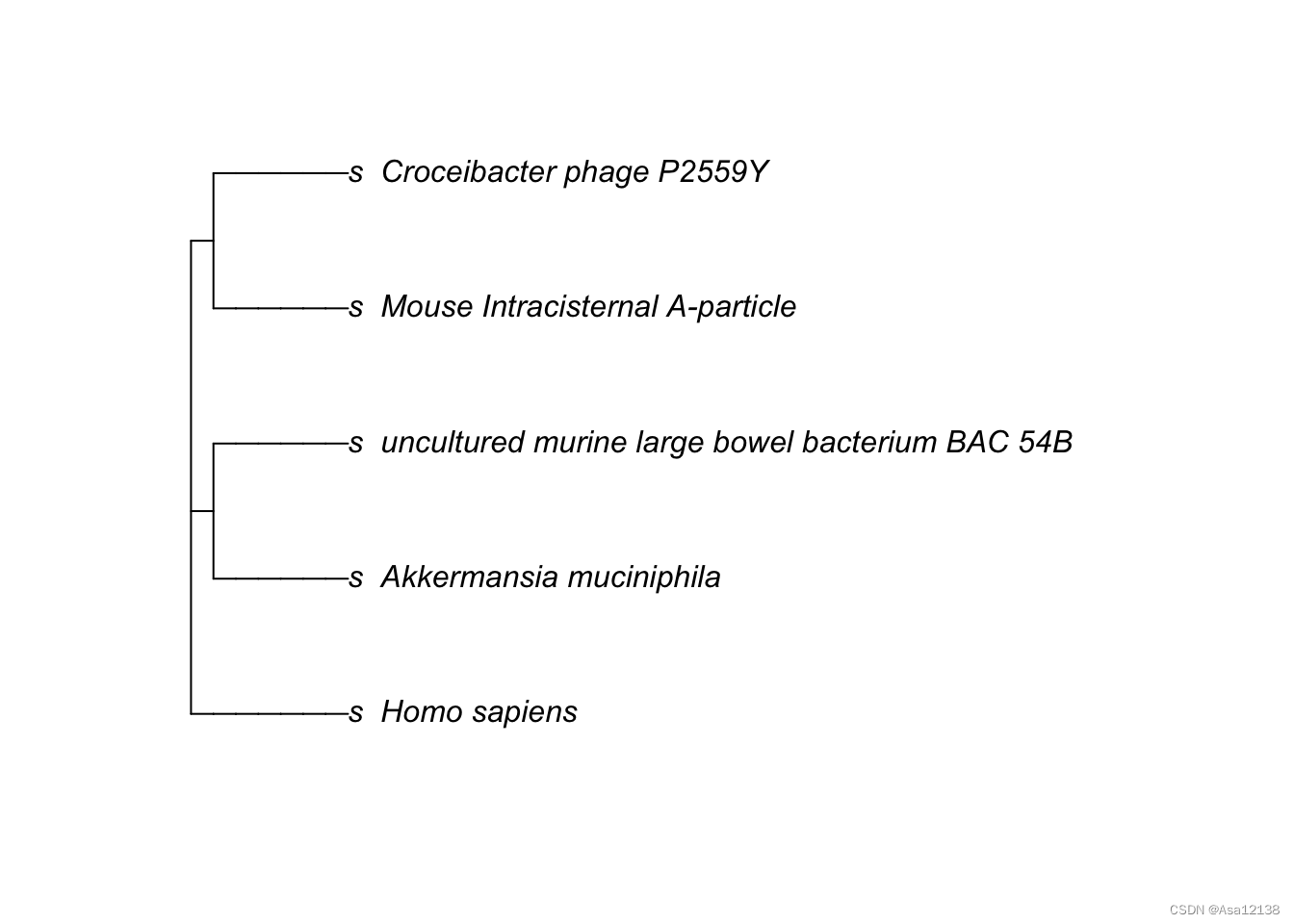
names <- system.file("extdata/name.txt", package = "pctax")%>%readLines()# 首先通过`name_or_id2df`获取整齐的系统发育分类:
tax_df=name_or_id2df(names,mode = "name")# 去除部分NA,原因可能是学名不标准,或者在新数据库里删除了,因为taxonomy数据库是不断变化的
tax_df=na.omit(tax_df)#用`df2tree`将分类层级表转化为树对象
tax_tree=pctax::df2tree(tax_df[,3:9])# tax_tree是phylo对象,可以用ape包直接简单绘图
ape:::plot.phylo(tax_tree)
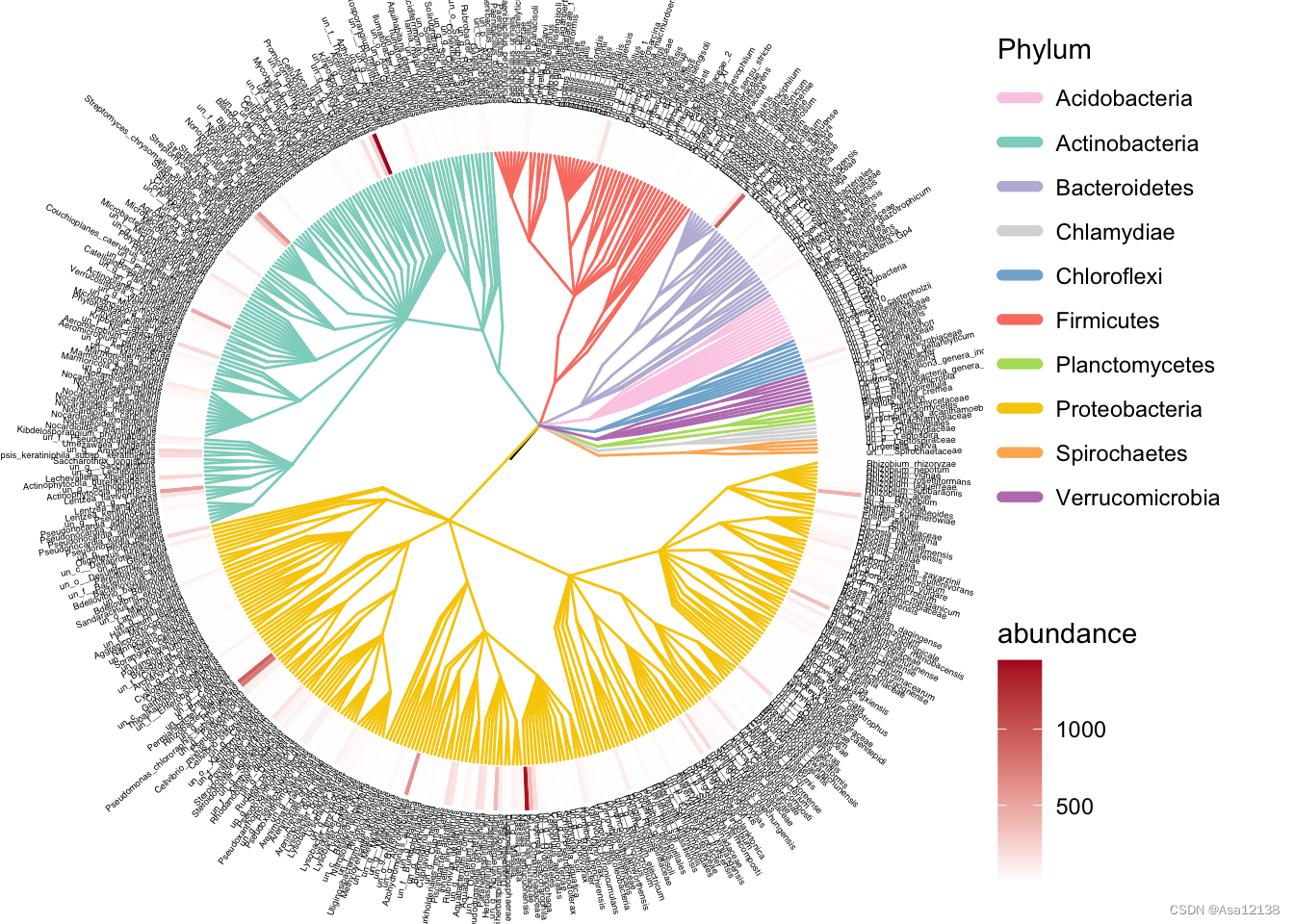
可视化
pctax还提供了一些系统发育信息展示方法:
- 系统发育树
data(otutab, package = "pcutils")
#otutab是丰度数据,taxonomy是分类层级表(可通过name_or_id2df获得)
ann_tree(taxonomy, otutab) -> treeeasy_tree(tree, add_abundance = TRUE) -> p
p
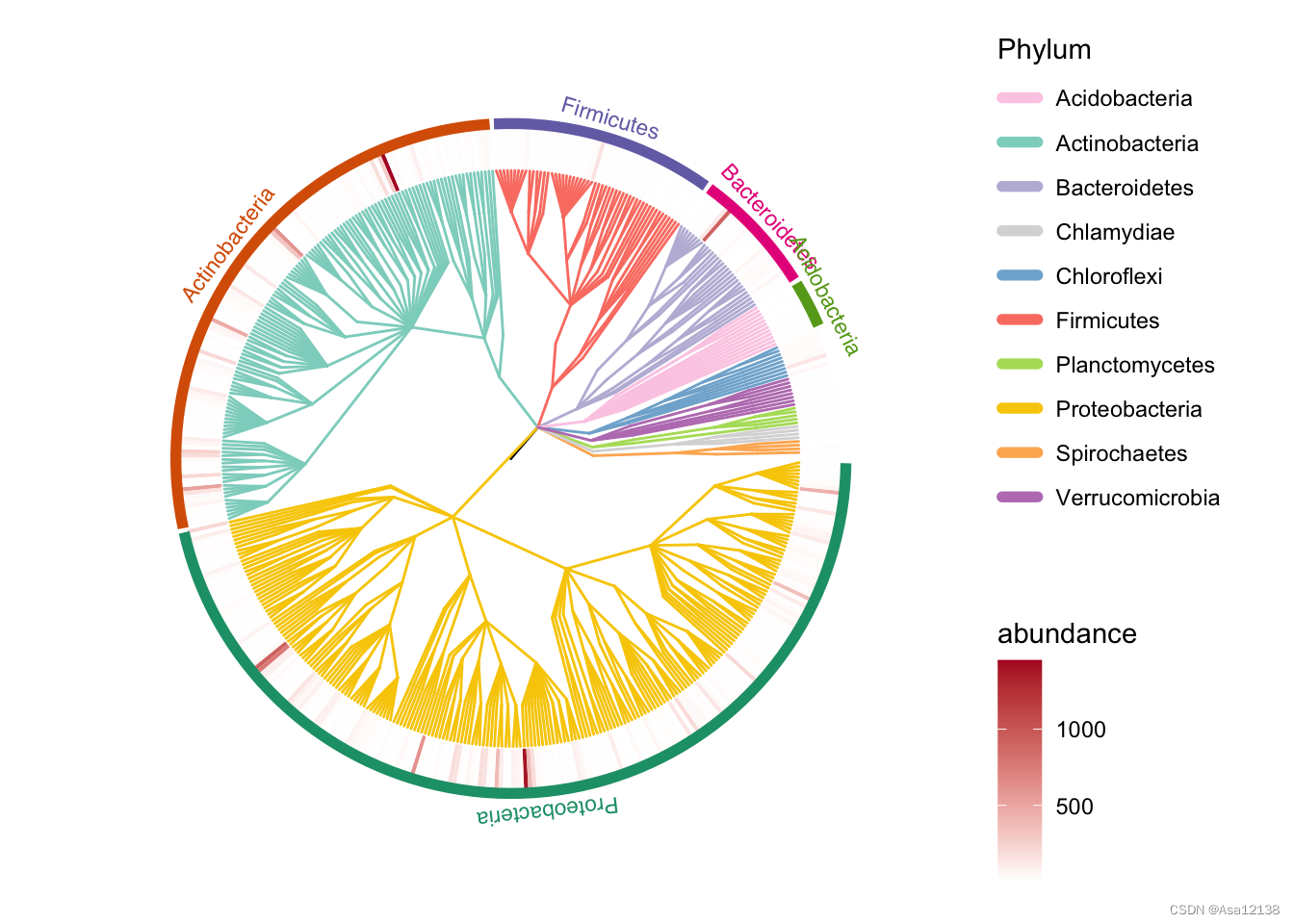
添加主要Phylum的strip:
easy_tree(tree, add_abundance = TRUE,add_tiplab = FALSE) -> p
some_tax <- table(taxonomy$Phylum) %>%sort(decreasing = TRUE) %>%head(5) %>%names()
add_strip(p, some_tax)
当然,更多系统发育树的绘制可以参考我之前写的R绘制优美的进化树(基础)和R绘制优美的进化树(进阶),或者使用iPhylo网站来交互式绘图:iPhylo 生成并绘制优美的分类树
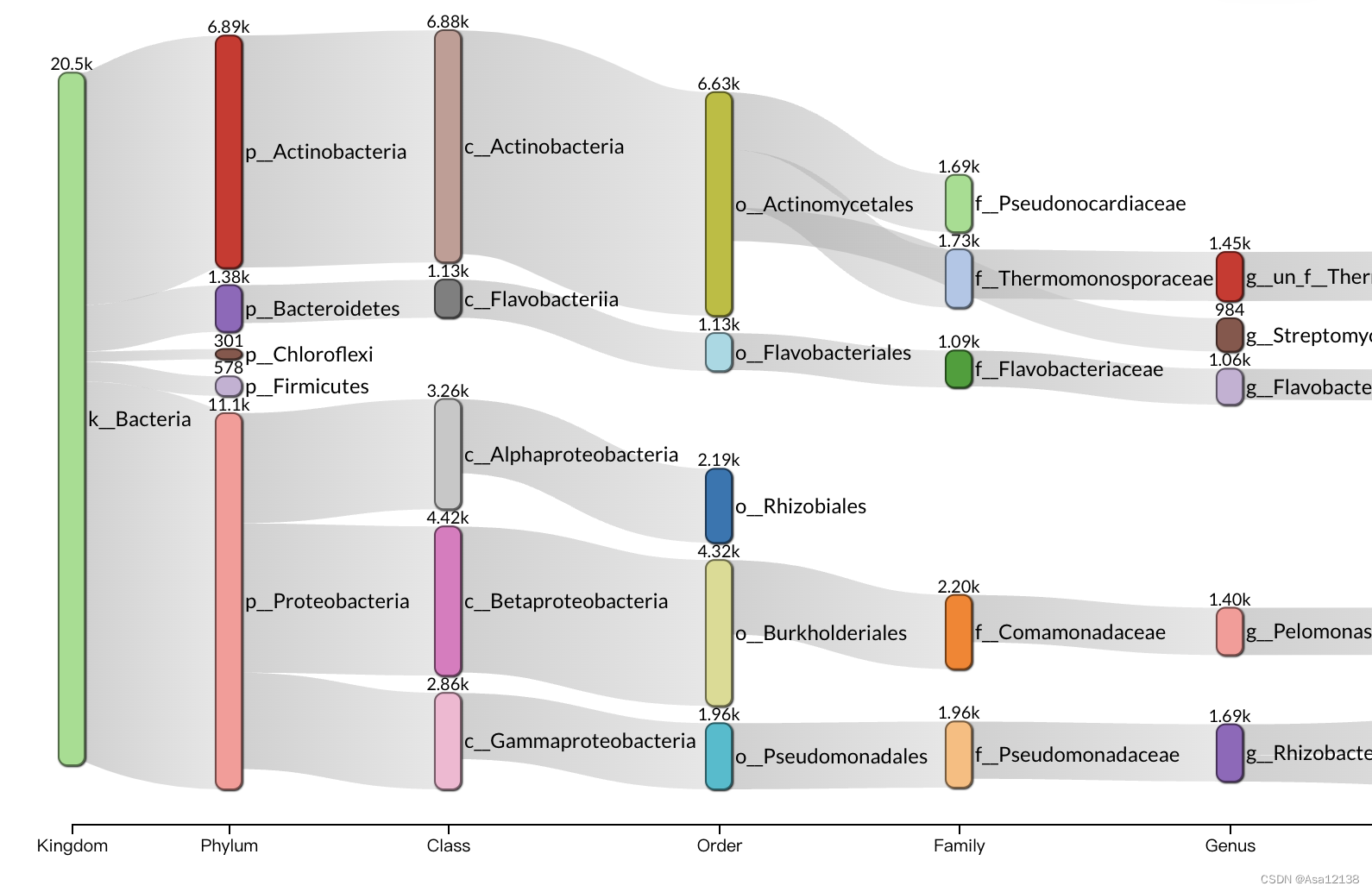
- 桑基图:
sangji_plot(tree)
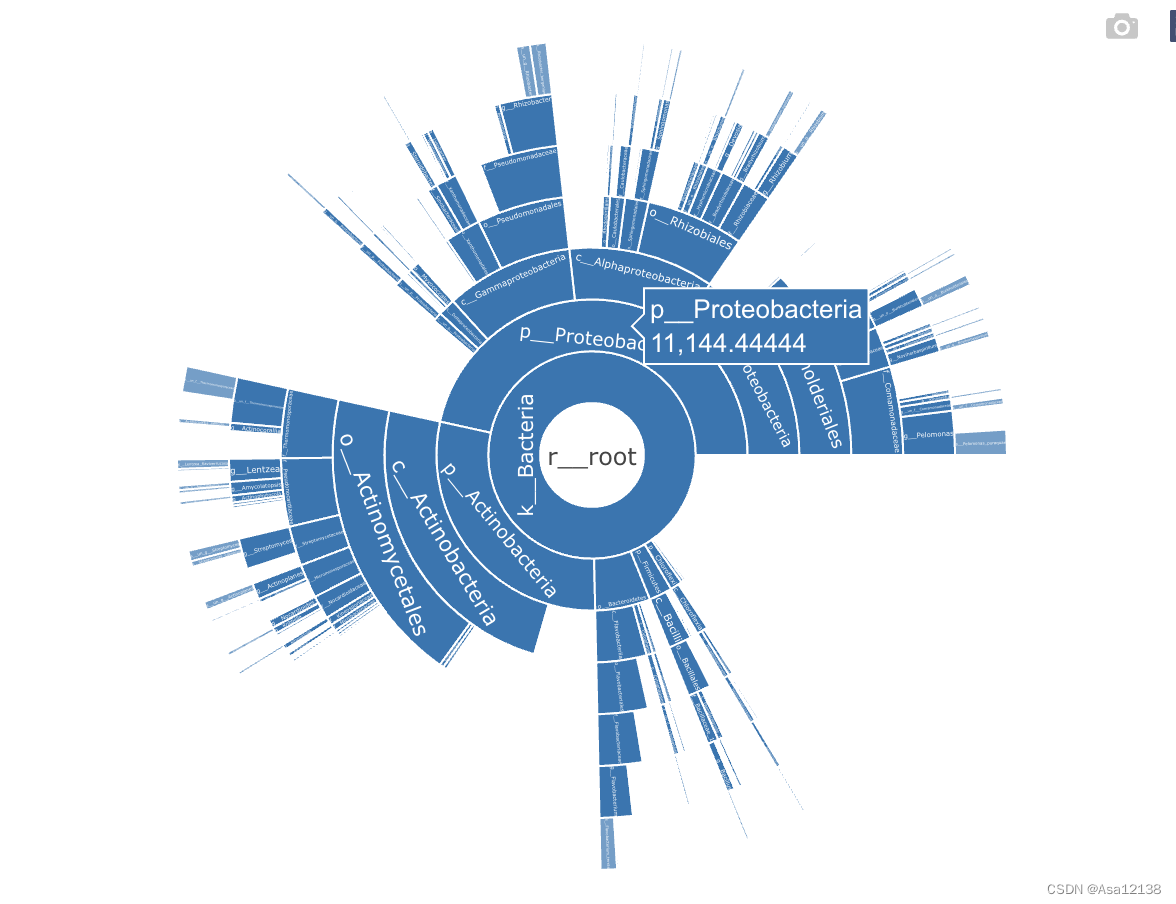
3.旭日图
sunburst(tree)
TaxonKit 使用
TaxonKit是采用Go语言编写的命令行工具, 提供Linux, Windows, macOS操作系统不同架构(x86-64/arm64)的静态编译的可执行二进制文件。
发布的压缩包不足3Mb,除了Github托管外,还提供国内镜像供下载,同时还支持conda和homebrew安装。
用户只需要下载、解压,开箱即用,无需配置,仅需下载解压NCBI Taxonomy数据文件解压到指定目录即可。
- 源代码 https://github.com/shenwei356/taxonkit ,
- 文档 http://bioinf.shenwei.me/taxonkit (介绍、使用说明、例子、教程)
选择系统对应的版本下载最新版 https://github.com/shenwei356/taxonkit/releases ,解压后添加环境变量即可使用。或可选conda安装
conda install taxonkit -c bioconda -y
表格数据处理,推荐使用 csvtk 更高效:
conda install csvtk -c bioconda -y
测试数据下载可直接 https://github.com/shenwei356/taxonkit 下载项目压缩包,或使用git clone下载项目文件夹,其中的example为测试数据
git clone https://github.com/shenwei356/taxonkit
TaxonKit为命令行工具,采用子命令的方式来执行不同功能, 大多数子命令支持标准输入/输出,便于使用命令行管道进行流水作业, 轻松整合进分析流程中。
- 输出:
- 所有命令输出中包含输入数据内容,在此基础上增加列。
- 所有命令默认输出到标准输出(stdout),可通过重定向(
>)写入文件。 - 或通过全局参数
-o或--out-file指定输出文件,且可自动识别输出文件后缀(.gz)输出gzip格式。
- 输入:
- 除了
list与taxid-changelog之外,lineage,reformat,name2taxid,filter与lca均可从标准输入(stdin)读取输入数据,也可通过位置参数(positional arguments)输入,即命令后面不带 任何flag的参数,如taxonkit lineage taxids.txt - 输入格式为单列,或者制表符分隔的格式,输入数据所在列用
-i或--taxid-field指定。
- 除了
TaxonKit直接解析NCBI Taxonomy数据文件(2秒左右),配置更容易,也便于更新数据,占用内存在500Mb-1.5G左右。 数据下载:
# 有时下载失败,可多试几次;或尝试浏览器下载此链接
wget -c https://ftp.ncbi.nih.gov/pub/taxonomy/taxdump.tar.gz
tar -zxvf taxdump.tar.gz# 解压文件存于家目录中.taxonkit/,程序默认数据库默认目录
mkdir -p $HOME/.taxonkit
cp names.dmp nodes.dmp delnodes.dmp merged.dmp $HOME/.taxonkit
Taxonkit的作者大大贴心地提供了中文文档:https://bioinf.shenwei.me/taxonkit/chinese/,非常详细,大家可以参考使用。

关注公众号 ‘bio llbug’,获取最新推送。
这篇关于R调用Taxonkit展示系统发育信息的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!



