本文主要是介绍④单细胞学习-cellchat细胞间通讯,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
目录
1,原理基础
流程
受体配体概念
方法比较
计算原理
2,数据
3,代码运行
1,原理基础
原文学习Inference and analysis of cell-cell communication using CellChat - PMC (nih.gov)
GitHub - sqjin/CellChat: R toolkit for inference, visualization and analysis of cell-cell communication from single-cell data
流程
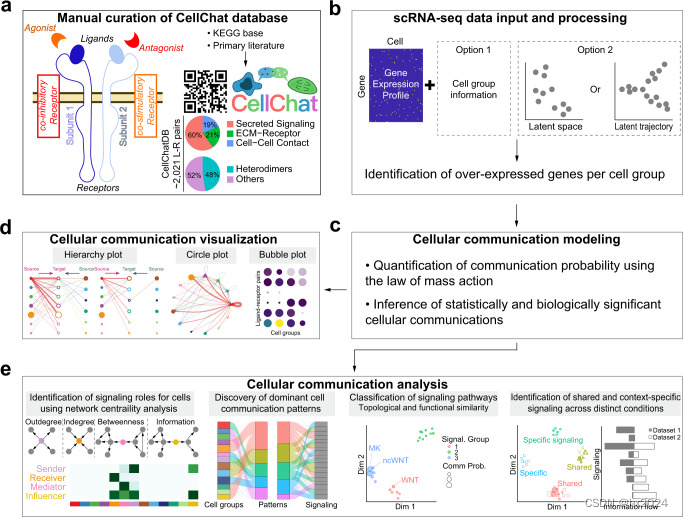
a 配体-受体相互作用数据库概述。CellChatDB考虑了配体-受体复合物的已知组成,包括具有多聚体配体和受体的复合物,以及几种辅因子类型:可溶性激动剂,拮抗剂,共刺激和共抑制膜结合受体。CellChatDB 包含 2021 个经过验证的交互,包括 60% 的分泌交互。此外,48%的相互作用涉及异聚分子复合物。b CellChat 要么要求用户分配单元格标签作为输入,要么根据作为输入提供的低维数据表示自动对单元格进行分组。c CellChat对通信概率进行建模,并识别重要的通信。d CellChat为不同的分析任务提供了多种可视化输出。层次图和圆图中的不同颜色表示不同的像元组。气泡图中的颜色与通信概率成正比,其中深色和黄色对应于最小和最大值。e CellChat通过图论、模式识别和流形学习等方法定量测量网络,以更好地促进细胞间通信网络的解释和设计原则的识别。除了分析单个数据集外,CellChat还描绘了不同背景下的信号变化,例如不同的发育阶段和生物条件。
受体配体概念
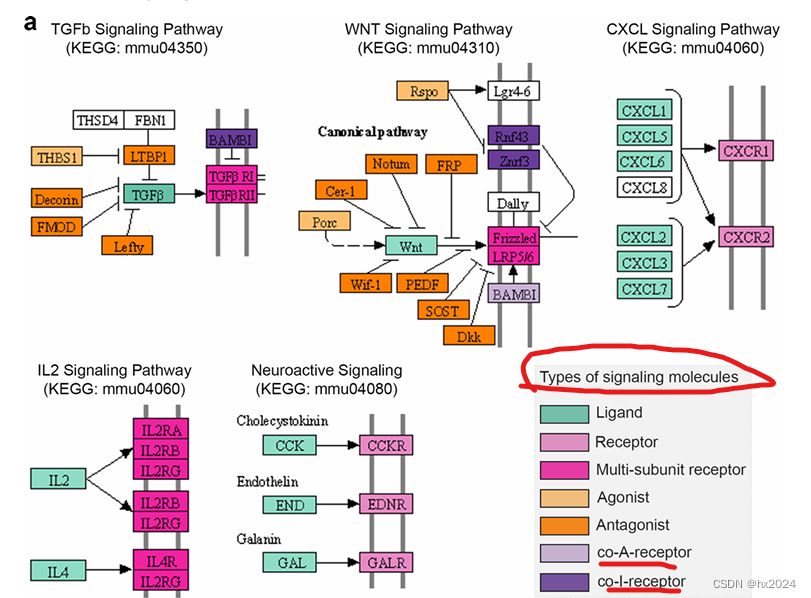
方法比较
- iTALK和NicheNet用于在scRNA-seq数据中推断细胞间的通讯关系,但这些方法只使用一个配体/受体基因对,常常忽略了许多受体中的多亚基复合物的作用。(例如,来自TGFβ途径的可溶性配体通过I型和II型受体的异聚复合物发出信号。),
- CellPhoneDB v2.0 可以预测两个细胞群之间的信号相互作用,解决了上述问题。但是,并未考虑其他重要的信号辅助因子,包括可溶性激动剂,拮抗剂以及刺激性和抑制性的膜结合型共受体。
CellChat细胞通讯分析(上)--文献解读 - 知乎 (zhihu.com)
文献阅读:使用 CellChat 推理和分析细胞-细胞通信 (qq.com)
计算原理
a) 鉴定差异表达的信号转导基因。为了推断细胞状态特异性通讯,我们首先使用显著性水平为 0.05 的 Wilcoxon 秩和检验,在给定的 scRNA-seq 数据集中鉴定了所有细胞群中差异表达的信号转导基因。
b) 集合平均表达式的计算。为了解释噪声效应,我们使用统计稳健的平均方法计算了给定细胞组中信号转导基因的集合平均表达。
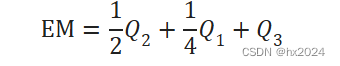
c) 细胞间通讯概率的计算。
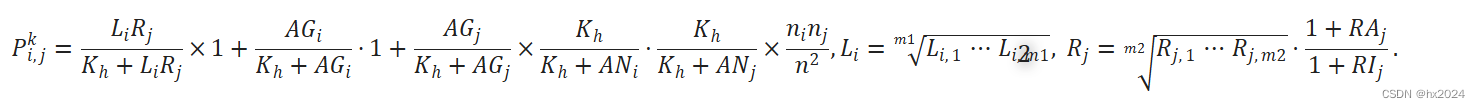
d) 鉴定具有统计学意义的细胞间通讯。通过随机排列细胞的组标签,然后重新计算通信概率P,使用排列检验来识别两个细胞组之间的显着相互作用。

cellchat的原理、cellchat能做哪些分析_哔哩哔哩_bilibili
2,数据
scRNA-seq data of human skin from patients with atopic dermatitis (figshare.com)
官方学习:focuslyj/CellChat - 码云 - 开源中国 (gitee.com)
tutorial · focuslyj/CellChat - 码云 - 开源中国 (gitee.com)
这个教程概述了使用 CellChat 对单个数据集进行细胞间通信网络的推理、分析和可视化的步骤。我们通过将 CellChat 应用于来自患者的病变(LS,患病)人类皮肤细胞的 scRNA-seq 数据来展示 CellChat 的多种功能。
3,代码运行
rm(list=ls())
library(CellChat)
library(patchwork)
library(ggplot2)
library(Seurat)
library(ggalluvial)#绘制桑基图
library(expm)
library(sna)
library(NMF)
options(stringsAsFactors = FALSE)##输入数据不自动转换成因子(防止数据格式错误)#数据:基因为行名,细胞为列名的基因表达矩阵(注意相关网络原因)
load(url("https://ndownloader.figshare.com/files/25950872")) #示例数据(正常+疾病)
#save(data_humanSkin,file = c("data_humanSkin.Rdata"))
rm(list=ls())
load("data_humanSkin.Rdata")
#数据查看
data.input = data_humanSkin$data # 提取标准化矩阵
meta = data_humanSkin$meta # a dataframe with rownames containing cell mata data
cell.use = rownames(meta)[meta$condition == "LS"] ##疾病组
#数据准备
data.input = data.input[, cell.use]
meta = meta[cell.use, ]
unique(meta$labels) # 检查细胞标签
#创建CellChat 对象
cellchat <- createCellChat(object = data.input, meta = meta, group.by = "labels")
cellchat <- addMeta(cellchat, meta = meta)#添加细胞信息
cellchat <- setIdent(cellchat, ident.use = "labels") # set "labels" as default cell identity
levels(cellchat@idents) # show factor levels of the cell labels
groupSize <- as.numeric(table(cellchat@idents)) # number of cells in each cell group#配体受体交互数据库查看
CellChatDB <- CellChatDB.human # use CellChatDB.mouse if running on mouse data
showDatabaseCategory(CellChatDB)
dplyr::glimpse(CellChatDB$interaction)
CellChatDB.use <- subsetDB(CellChatDB, search = "Secreted Signaling") # 分泌信号
cellchat@DB <- CellChatDB.use #在对象中设置使用的数据库
#预处理用于细胞通信分析的表达数据
cellchat <- subsetData(cellchat)
#future::plan("multiprocess", workers = 4) #可以不运行
cellchat <- identifyOverExpressedGenes(cellchat)#分析差异基因
cellchat <- identifyOverExpressedInteractions(cellchat)#
cellchat <- projectData(cellchat, PPI.human)#进行细胞通信网络的推断
cellchat <- computeCommunProb(cellchat, raw.use = TRUE)#计算通信概率并推断cellchat网络
cellchat <- filterCommunication(cellchat, min.cells = 10)#过滤
#信号通路级别推断细胞-细胞通信
cellchat <- computeCommunProbPathway(cellchat)
#计算整合的细胞通信网络
cellchat <- aggregateNet(cellchat)#进行可视化
groupSize <- as.numeric(table(cellchat@idents))
par(mfrow = c(1,2), xpd=TRUE)
netVisual_circle(cellchat@net$count,vertex.weight = groupSize, weight.scale = T, label.edge= F, title.name = "Number of interactions")
netVisual_circle(cellchat@net$weight, vertex.weight = groupSize,weight.scale = T, label.edge= F, title.name = "Interaction weights/strength")mat <- cellchat@net$weight
par(mfrow = c(3,4), xpd=TRUE)##如果可视化出现问题可以把前面的图片保存清空后再作图
for (i in 1:nrow(mat)) {mat2 <- matrix(0, nrow = nrow(mat), ncol = ncol(mat), dimnames = dimnames(mat))mat2[i, ] <- mat[i, ]netVisual_circle(mat2, vertex.weight = groupSize, weight.scale = T, edge.weight.max = max(mat), title.name = rownames(mat)[i])
}
dev.off()#细胞通信网络的可视化######################################
pathways.show <- c("CXCL")
vertex.receiver = seq(1,4) # a numeric vector.
netVisual_aggregate(cellchat, signaling = pathways.show,vertex.receiver = vertex.receiver)# Circle plot
par(mfrow=c(1,1))
netVisual_aggregate(cellchat, signaling = pathways.show, layout = "circle")# 弦图
par(mfrow=c(1,1))
netVisual_aggregate(cellchat, signaling = pathways.show, layout = "chord")#热图
par(mfrow=c(1,1))
netVisual_heatmap(cellchat, signaling = pathways.show, color.heatmap = "Reds")# Chord diagram
group.cellType <- c(rep("FIB", 4), rep("DC", 4), rep("TC", 4)) # grouping cell clusters into fibroblast, DC and TC cells
names(group.cellType) <- levels(cellchat@idents)
netVisual_chord_cell(cellchat, signaling = pathways.show,group = group.cellType, title.name = paste0(pathways.show, " signaling network"))#计算每个配体受体对整体信号通路的贡献,并可视化由单个配体受体对调节的细胞通信
netAnalysis_contribution(cellchat, signaling = pathways.show)#视化由单个配体受体对调节的细胞-细胞通信
pairLR.CXCL <- extractEnrichedLR(cellchat, signaling = pathways.show, geneLR.return = FALSE)
LR.show <- pairLR.CXCL[1,] # show one ligand-receptor pair
vertex.receiver = seq(1,4) # # Hierarchy plot a numeric vector
netVisual_individual(cellchat, signaling = pathways.show, pairLR.use = LR.show, vertex.receiver = vertex.receiver)# Circle plot
netVisual_individual(cellchat, signaling = pathways.show, pairLR.use = LR.show, layout = "circle")#相关保存
pathways.show.all <- cellchat@netP$pathways
# check the order of cell identity to set suitable vertex.receiver
levels(cellchat@idents)
vertex.receiver = seq(1,4)
for (i in 1:length(pathways.show.all)) {# Visualize communication network associated with both signaling pathway and individual L-R pairsnetVisual(cellchat, signaling = pathways.show.all[i], vertex.receiver = vertex.receiver, layout = "hierarchy")# Compute and visualize the contribution of each ligand-receptor pair to the overall signaling pathwaygg <- netAnalysis_contribution(cellchat, signaling = pathways.show.all[i])ggsave(filename=paste0(pathways.show.all[i], "_L-R_contribution.pdf"), plot=gg, width = 3, height = 2, units = 'in', dpi = 300)
}#可视化由多个配体受体或信号通路调节的细胞通信
netVisual_bubble(cellchat, sources.use = 4, targets.use = c(5:11), remove.isolate = FALSE)
netVisual_bubble(cellchat, sources.use = 4, targets.use = c(5:11), signaling = c("CCL","CXCL"), remove.isolate = FALSE)netVisual_chord_gene(cellchat, sources.use = 4, targets.use = c(5:11), lab.cex = 0.5,legend.pos.y = 30)
# show all the interactions received by Inflam.DC
netVisual_chord_gene(cellchat, sources.use = c(1,2,3,4), targets.use = 8, legend.pos.x = 15)#使用小提琴/点图绘制信号基因表达分布
plotGeneExpression(cellchat, signaling = "CXCL")
plotGeneExpression(cellchat, signaling = "CXCL", enriched.only = FALSE)#提取与推断的L-R对或信号通路相关的信号基因#细胞通信网络系统分析############################
cellchat <- netAnalysis_computeCentrality(cellchat, slot.name = "netP") # the slot 'netP' means the inferred intercellular communication network of signaling pathways
netAnalysis_signalingRole_network(cellchat, signaling = pathways.show, width = 8, height = 2.5, font.size = 10)
#使用散点图在 2D 空间中可视化占主导地位的发射器(源)和接收器(目标)
gg1 <- netAnalysis_signalingRole_scatter(cellchat)
gg2 <- netAnalysis_signalingRole_scatter(cellchat, signaling = c("CXCL", "CCL"))
gg1 + gg2#哪些信号对某些细胞组的传出或传入信号贡献最大
ht1 <- netAnalysis_signalingRole_heatmap(cellchat, pattern = "outgoing")
ht2 <- netAnalysis_signalingRole_heatmap(cellchat, pattern = "incoming")
ht1 + ht2ht <- netAnalysis_signalingRole_heatmap(cellchat, signaling = c("CXCL", "CCL"))#识别和可视化分泌细胞的传出通信模式##############################
selectK(cellchat, pattern = "outgoing")#selectK推断模式的数量。
#当传出模式数为 3 时,Cophenetic 和Silhouette值都开始突然下降。
nPatterns = 3
cellchat <- identifyCommunicationPatterns(cellchat, pattern = "outgoing", k = nPatterns)netAnalysis_dot(cellchat, pattern = "outgoing")#绘制点图
selectK(cellchat, pattern = "incoming")#识别和可视化目标细胞的传入通信模式
nPatterns = 4
cellchat <- identifyCommunicationPatterns(cellchat, pattern = "incoming", k = nPatterns)# river plot桑基图
library(ggalluvial)
netAnalysis_river(cellchat, pattern = "incoming")
# dot plot
netAnalysis_dot(cellchat, pattern = "outgoing")#信号网络的流形和分类学习分析
cellchat <- computeNetSimilarity(cellchat, type = "functional")
cellchat <- netEmbedding(cellchat, type = "functional")
#> Manifold learning of the signaling networks for a single dataset
cellchat <- netClustering(cellchat, type = "functional")
#> Classification learning of the signaling networks for a single dataset
# Visualization in 2D-space
netVisual_embedding(cellchat, type = "functional", label.size = 3.5)#最后保存
saveRDS(cellchat, file = "cellchat_humanSkin_LS.rds")
4,流程导图
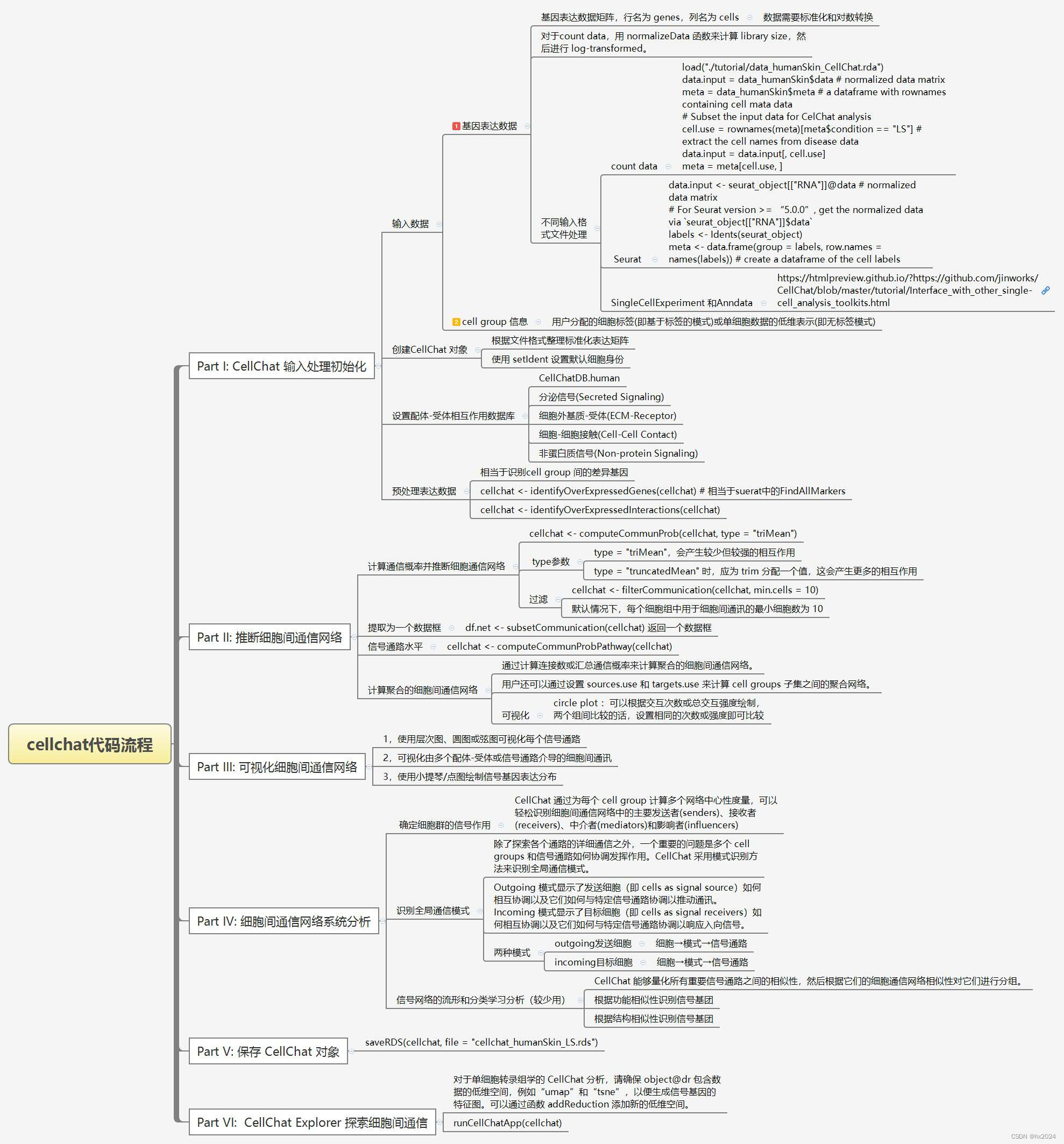
4程序员 - CellChat v2 教程 1:使用 CellChat 推理和分析细胞间通信 - 个人文章 - SegmentFault 思否
参考:
1:Inference and analysis of cell-cell communication using CellChat
这篇关于④单细胞学习-cellchat细胞间通讯的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!