本文主要是介绍转录组总结,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
1. 软件安装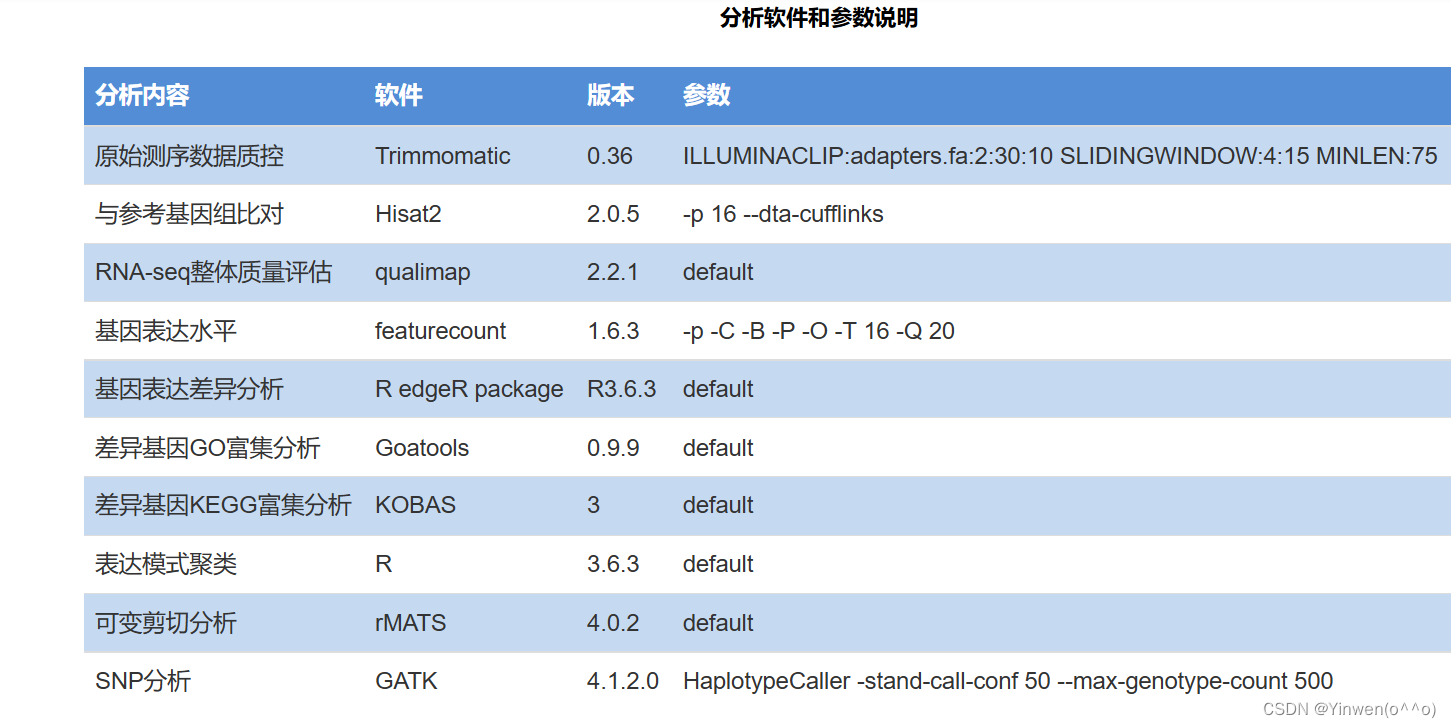
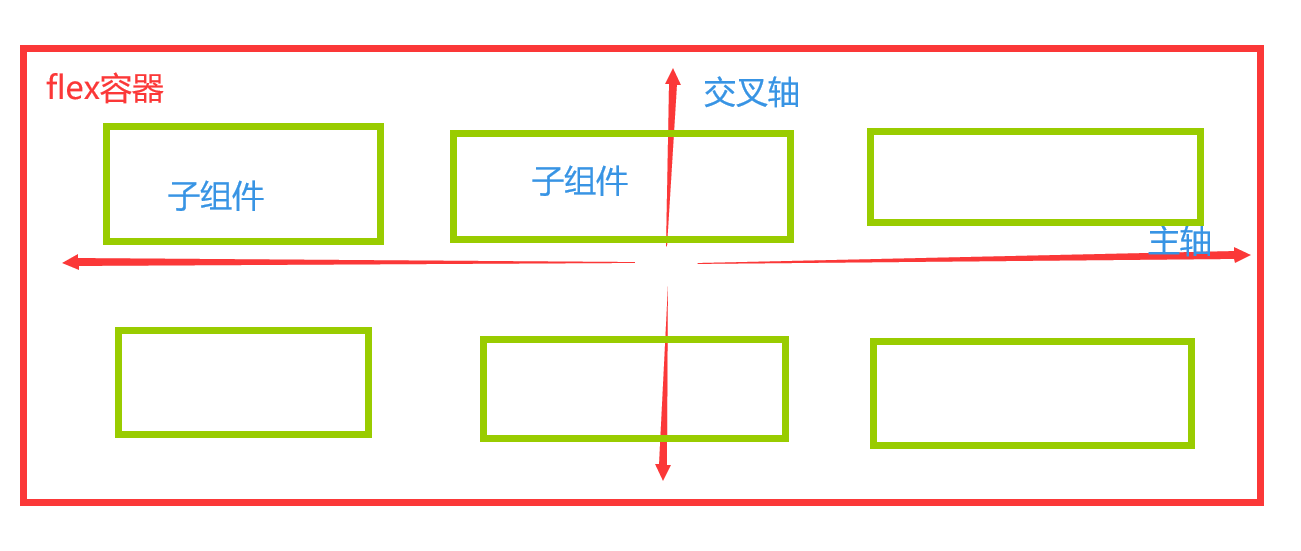
2.转录组分析步骤:
① 建立环境
#建立python2.7的环境,大部分的转录组信息都需要在Python2的环境下进行
conda create -n py2env python=2.7
source activate py2env② 获取fastqc报告
#单个报告
fastqc -t 15 /home/yinwen/biosoft/DNG_part/DG5_1_R1.fq.gz -o ~/#批量报告
fastqc -t 15 /home/yinwen/biosoft/DNG_part/*fq.gz -o /home/yinwen/RNA-seq_report/#打包报告
tar -cvf html_files.tar *.html③ 获取综合的 Multiqc 报告
#将fastqc生成的多个报告整合成一个报告,方便查看所有测序数据的质量
multiqc .④ 分析报告
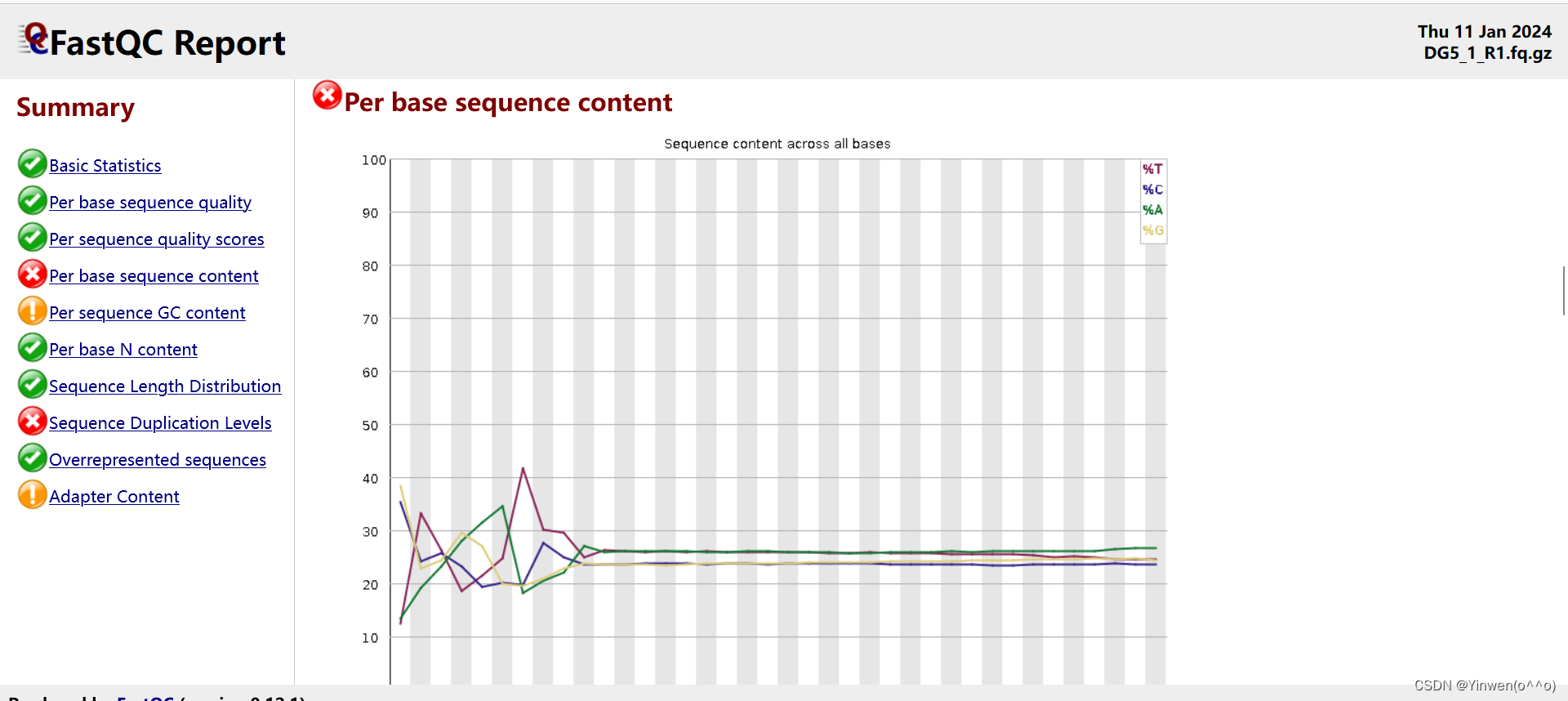
我的报告:
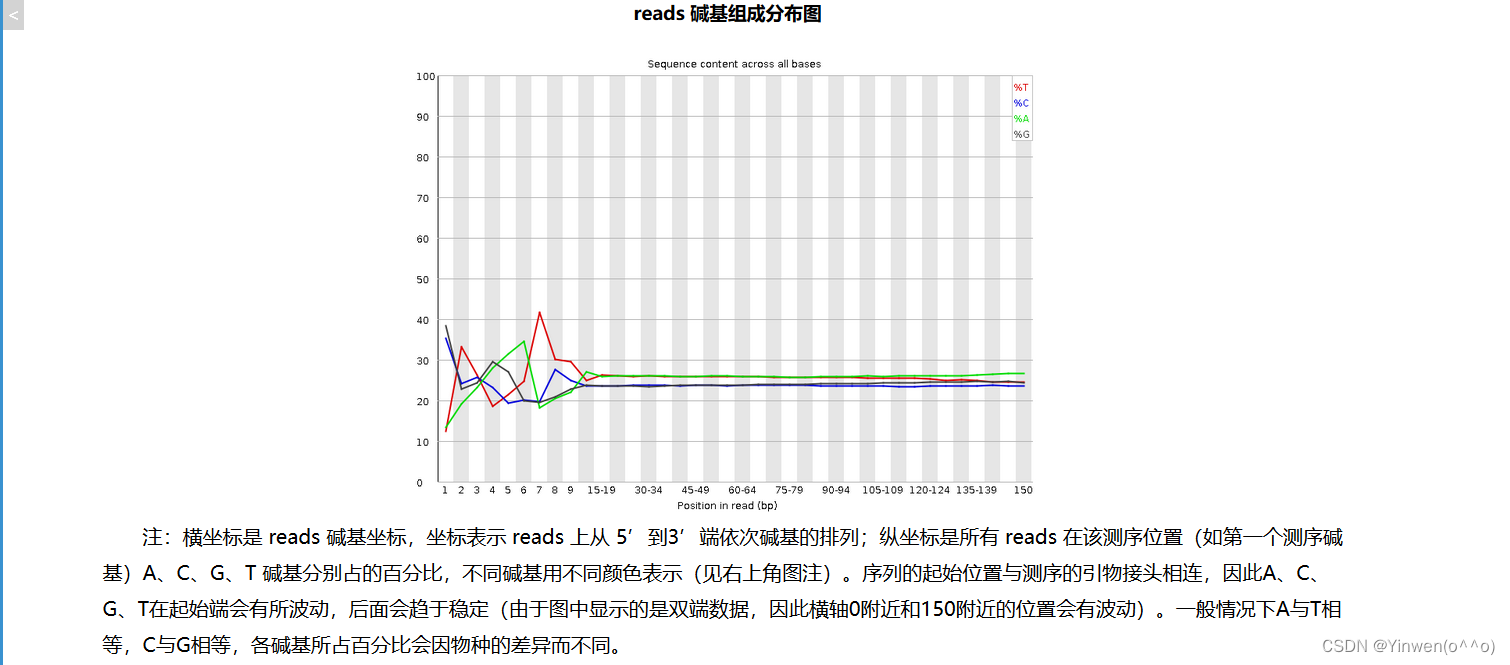
公司报告:
#trimmomatic修饰参数
trimmomatic PE -phred33 DG5_1_R1.fq.gz DG5_1_R2.fq.gz output_forward1__paired.fq.gz output_forward1_unpaired.fq.gz output_reverse2_paired.fq.gz output_reverse2_unpaired.fq.gz ILLUMINACLIP:/home/yinwen/miniconda3/pkgs/trimmomatic-0.39-hdfd78af_2/share/trimmomatic-0.39-2/adapters/TruSeq3-PE.fa:2:30:10 LEADING:3 TRAILING:20 SLIDINGWINDOW:4:15 HEADCROP:18 MINLEN:75
#公司参数
ILLUMINACLIP:adapters.fa:2:30:10 SLIDINGWINDOW:4:15 MINLEN:75⑤ 建立索引
# 重命名后构建索引
hisat2-build dananguo_genome.fa genome(py2env) [yinwen@node hisat2]$ ls -l
total 765852
-rw-rw-r-- 1 yinwen yinwen 174481544 Jan 27 16:29 genome.1.ht2
-rw-rw-r-- 1 yinwen yinwen 127714808 Jan 27 16:29 genome.2.ht2
-rw-rw-r-- 1 yinwen yinwen 287 Jan 27 16:18 genome.3.ht2
-rw-rw-r-- 1 yinwen yinwen 127714802 Jan 27 16:18 genome.4.ht2
-rw-rw-r-- 1 yinwen yinwen 224243805 Jan 27 16:31 genome.5.ht2
-rw-rw-r-- 1 yinwen yinwen 130051792 Jan 27 16:31 genome.6.ht2
-rw-rw-r-- 1 yinwen yinwen 12 Jan 27 16:18 genome.7.ht2
-rw-rw-r-- 1 yinwen yinwen 8 Jan 27 16:18 genome.8.ht2⑥ 进行比对
#运行后得到.sam文件
hisat2 -x genome -p 5 -1 /home/yinwen/clean/DG5_1_R1_val_1.fq -2 /home/yinwen/clean/DG5_1_R2_val_2.fq -S genome.sam
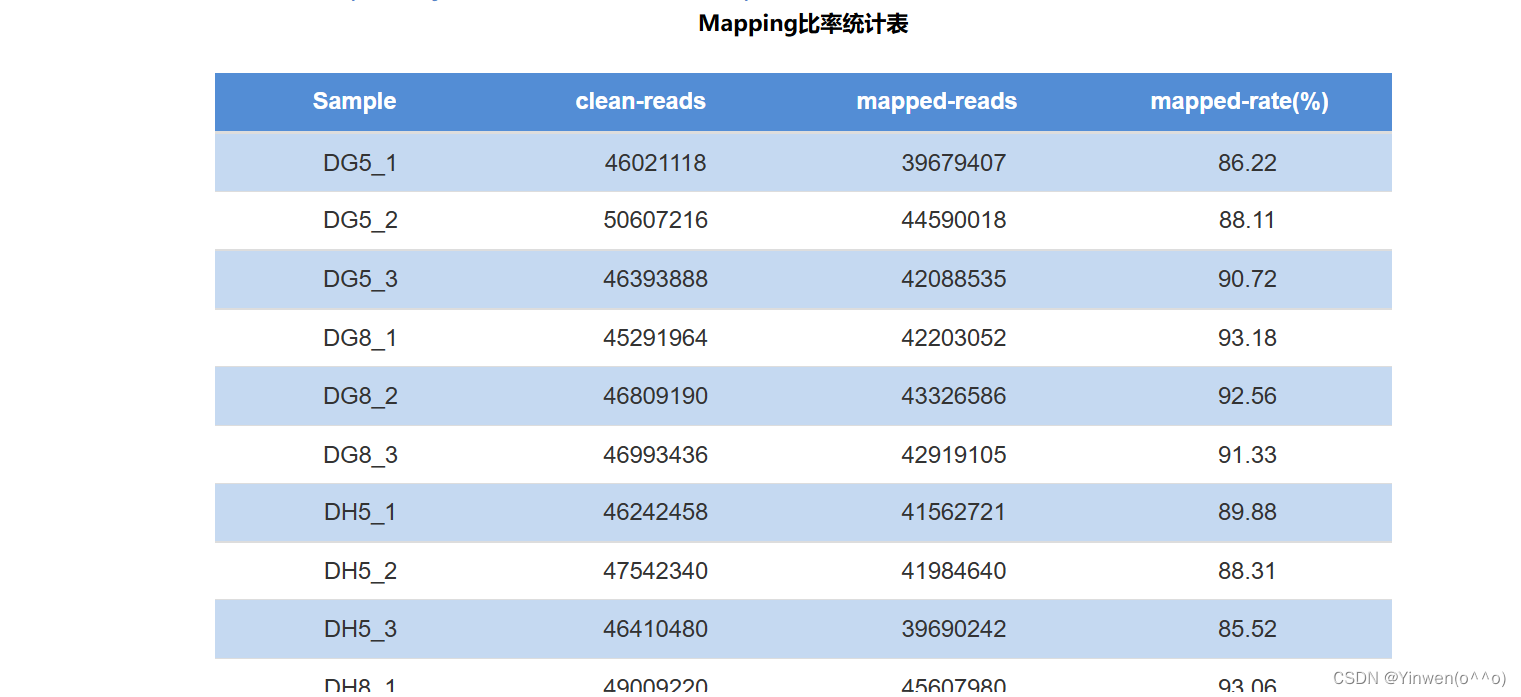
#公司与参考基因组比对:
-p 16 --dta-cufflinks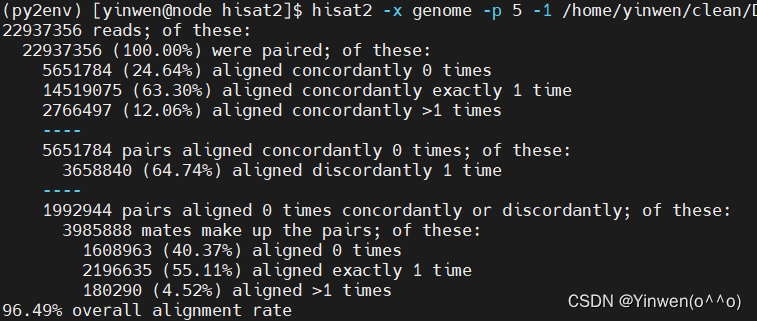
自己的数据比对率为96.49%,公司的比率比我们得到的比率要低,说明处理依旧需要细化。
⑦ 排序压缩
#压缩:把 sam文件 转为 bam文件
samtools view -S genome.sam -b > genome.bam
#排序:samtools sort
samtools sort -n -@ 5 genome.bam -o genome
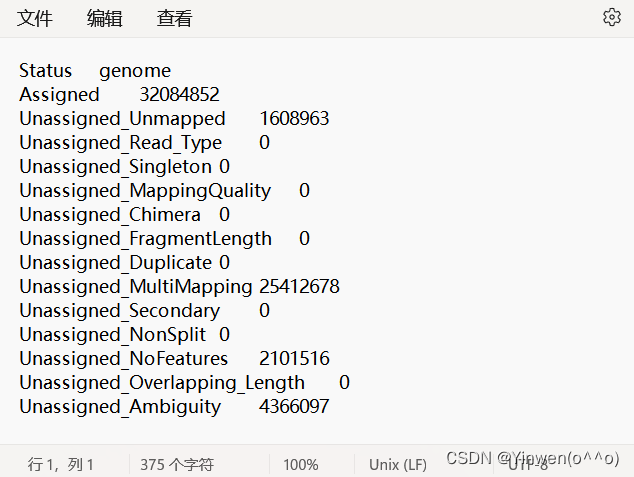
⑧ 计数统计
gene_name.counts.summary 文件是计数统计情况
gene_name.counts 文件是基因的具体信息
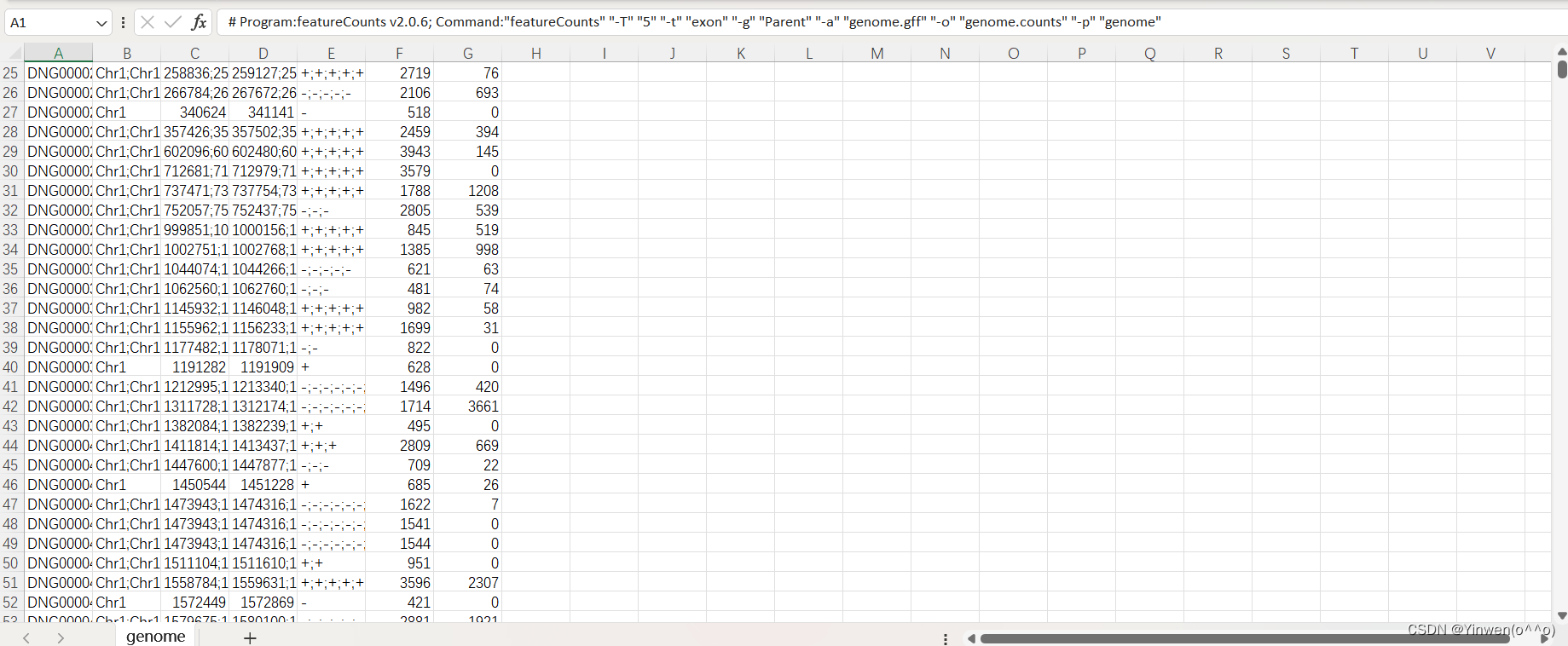
featureCounts -T 5 -t exon -g Parent -a genome.gff -o genome.counts -p genome
#公司其他参数
-p -C -B -P -O -T 16 -Q 20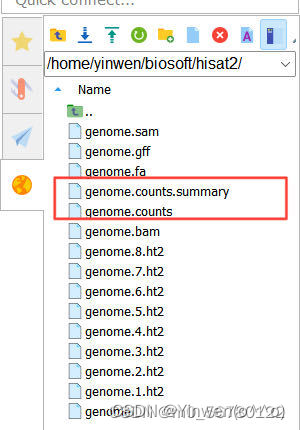
拓展:【生物信息】RPKM, FPKM和TPM
这篇关于转录组总结的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!