本文主要是介绍sc-MAVE,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
Deep-joint-learning analysis model of single cell transcriptome and open chromatin accessibility data单细胞转录组和开放染色质可及性数据的深度联合学习分析模型
在同一个细胞中同时分析转录组和染色质可及性信息为了解细胞状态提供了前所未有的解决方案。然而,计算有效的方法,这些固有的稀疏和异构数据的整合是缺乏的。在这里,我们提出了一个单细胞多模态变分自动编码器模型,它结合了三种类型的联合学习策略与概率高斯混合模型,以学习准确代表这些多层配置文件的联合潜在特征。对模拟数据集和真实的数据集的研究表明,该方法具有更好的能力:(i)在联合学习空间中剖析细胞异质性;(ii)去噪和估算数据;(iii)构建多层组学数据之间的关联,可用于理解转录调控机制。
介绍
基因表达是通过一组转录因子(TF)结合到其顺式调控基因组区域来调节的。
scRNA-seq表征细胞的基因表达水平,而scATAC-seq等表观基因组变化反映了附近基因中顺式调控元件的开放性。这种两组学数据的整合可以提供关于与细胞异质性相关的调控层的新见解[13]。许多集成工具都是为批量数据设计的[14]。
例如,主成分分析(PCA)的推广MOFA被提出来处理批量数据,也可以应用于单细胞数据集[15]。IntNMF是非负矩阵因子分解(NMF)的扩展,用于整合疾病亚型分类的多组学数据,并评估其处理单细胞数据集[16,17]。然而,最近的研究发现,单细胞数据有其独特的特点,不同于批量数据,因此需要开发新的方法[18]。单细胞多分析(PCA)的集成被提出来处理批量数据,也可以应用于单细胞数据集[15]。IntNMF是非负矩阵因子分解(NMF)的扩展,用于整合疾病亚型分类的多组学数据,并评估其处理单细胞数据集[16,17]。然而,最近的研究发现,单细胞数据有其独特的特点,不同于批量数据,因此需要开发新的方法[18]。
单细胞多组学数据的整合仍然是一个巨大的挑战,这是由于固有的高度稀疏性,由于测定噪声导致的巨大异质性,scATAC-seq和scRNA-seq数据之间的巨大维度差异,大约10-20倍[19],以及越来越大规模的数据集[20]。已经开发了大量用于scRNA-seq数据整合的方法,然而,只有少数方法被提出用于整合单细胞多组学数据,并且这些方法是针对从不同细胞收集但从相同细胞群体提取的组学数据开发的[21-24]。例如,提出了耦合MMF,通过构建基因和顺式调控元件的耦合非负矩阵来对scRNA-seq和scATACseq数据进行聚类[23]。MATCHER被提出来通过使用高斯过程潜变量模型来推断每个细胞的伪时间来预测scRNA-seq和scATACseq之间的相关性[24]。最近,开发了Seurat(版本3)[25]和LIGER [22]用于整合scRNA-seq和scATAC-seq数据。这两种方法都是先将scATAC-seq数据转化为类似于基因表达数据的基因活性数据,然后通过在低维空间中相互比对来识别scRNA-seq数据和基因活性数据之间的锚点。然而,两组学/两层组学数据之间的比对效率通常需要来自两种测量的相似聚类性能。由于scATAC-seq数据的极稀疏性(即sci-CAR-seq中超过99%为零),很难通过scATAC-seq数据定义细胞簇。因此,这两种方法的不正确对齐可能会影响下游分析。
深度生成模型已经成为一个强大的框架来建模高维数据[26,27]。具体地,VAE通过编码器从输入数据学习低维特征,并通过解码器恢复输入数据,这可以通过最大化恢复的数据和输入数据之间的似然性,并最小化学习的潜在特征和真实后验之间的Kullback-Leibler(KL)发散来完成。最近,提出了采用标准VAE的单细胞变分推理(scVI)来分析scRNA-seq数据[26]。然而,标准的VAE在潜在变量上使用单一的各向同性多变量高斯分布,并且通常不适合稀疏数据[28]。SCALE适配使用高斯混合模型(GMM)作为潜在变量的先验的VAE被提出来分析scATAC-seq数据,分析结果表明,集成VAE和GMM的框架可以用于处理高度稀疏的数据,并学习更分散和可解释的潜在特征[27]。深度学习多模态技术[29,30]的最近快速发展以及在整合多视图生物数据[31]方面的成功应用,证明了它们在解决当前单细胞多组学数据分析困难方面的巨大潜力。
在这里,我们提出了单细胞多模态变分自动编码器(scMVAE),用于整合来自同一单细胞的scRNAseq和scATAC-seq数据,通过使用三种类型的联合学习策略。scMVAE模型使用随机优化和多模态编码器,首先聚合两种组学数据跨相似细胞和特征,以逼近具有GMM先验的联合潜在特征位置,然后通过每种组学数据的解码器重构观察到的表达值,同时考虑每种类型数据的归一化,可用于训练非常大的数据集。
特别是,通过无监督方式联合学习两种组学数据,scMVAE模型(i)产生具有生物意义的低维特征,同时表示这两个多层剖面,允许细胞可视化和聚类;(ii)去噪和填充两种组学数据;(iii)构建两层数据之间的关联,可用于推断新的调控关系。为了证明其效率,我们将scMVAE模型和其他整合方法应用于模拟和真实数据集,结果表明scMVAE模型的性能优于当前的最先进方法。
方法
scMVAE概率模型
scMVAE通过三种联合学习策略对来自同一细胞的scRNA-seq和scATAC-seq的分布进行建模:PoE推断网络(在材料S1中详细描述)、神经网络和直接连接两种组学数据特征(图1A–C)。为了平衡scRNA-seq和scATAC-seq数据之间的大尺度差异,我们将scATAC-seq数据的峰值水平计数矩阵转换为类似于scRNA-seq数据的基因活性数据,建模每个组学数据均来自于一个零膨胀负二项(ZINB)分布。
具体而言,给定K个聚类,可以通过多组学编码器网络通过重新参数化获得联合学习特征z,其中c是一个概率离散的分类变量。p(z|c)是一个混合高斯分布,其参数是由在c条件下的均值向量μc和协方差矩阵σc参数化的。考虑到x、y和c在z条件下是独立的,那么多模态联合学习分布p(x、y、z、c、lx、ly),其中lx和ly分别是用作scRNA-seq和scATAC-seq数据的库大小因子的一维高斯变量,可以分解为:
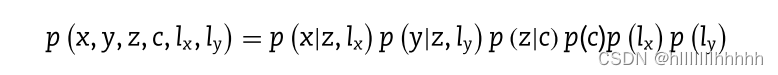


每个因子分解变量定义如下:
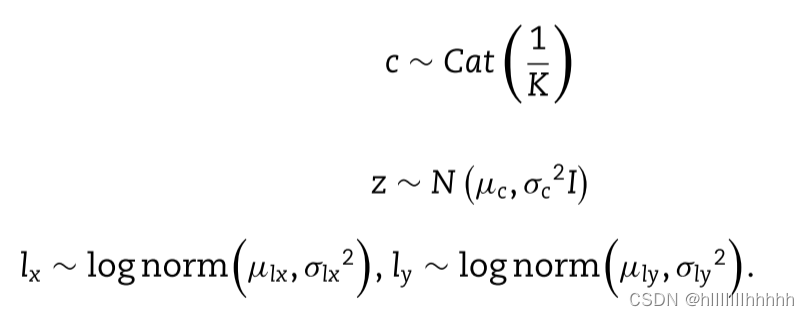
此外,x或y的每个基因表达水平独立于以下生成过程:
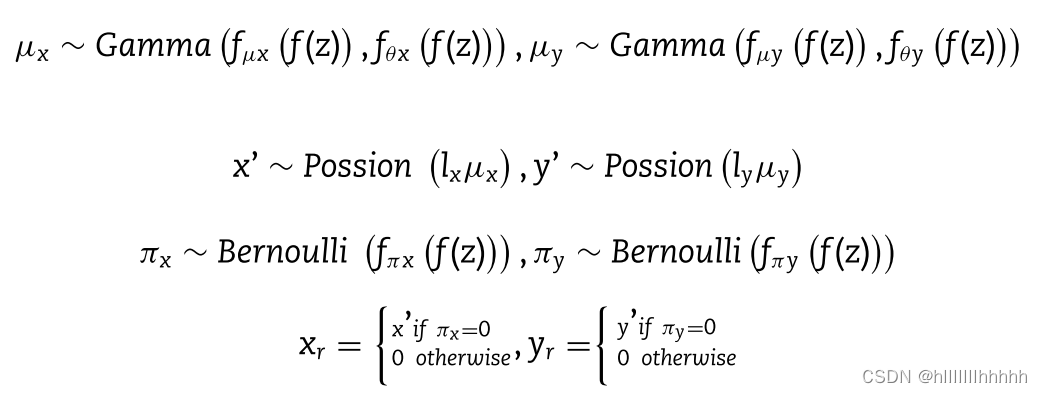
在MVAE中,z的GMM先验被用来生成高度逼真的样本,通过学习更加解耦和可解释的潜在表示。这在先前的工作中分别应用于scRNA-seq和scATAC-seq[27, 32]。lx和ly被视为与经验日志库大小强相关的对数正态分布。fθx(f(z))和fθy(f(z))表示由变分贝叶斯推断估计的特定特征的反比例。
在推断期间,神经网络fμx和fμy通过在最后一层使用‘softmax’激活函数被约束,以编码一个细胞中所有基因的平均比例基因表达,分别用于scRNA-seq和scATAC-seq数据。神经网络fπx和fπy通过在最后一层使用‘sigmoid’函数编码每个基因是否因为捕获效率和测序深度而被删除,用于每个二组学数据。
scMVAE模型的训练旨在最大化观察到的scRNA-seq和scATAC-seq数据的对数似然,然而,由于这是不可解的,因此转而优化证据下界(ELBO):
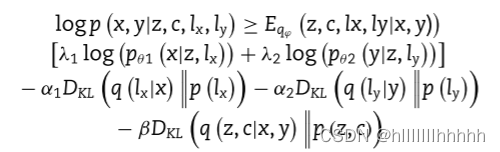
鼓励使用与库大小因子lx和ly相关的两个重建项和KL散度的正则化项来进行数据归一化、去噪和插值。潜在变量z的KL散度用于将其调节为GMM流形,以增强与多组学数据的关联。参数qϕ、pθ1和pθ2分别是多模态编码器、scRNA-seq数据的解码器和scATAC-seq数据的解码器。
所有神经网络都使用了dropout正则化和批量归一化。每个神经网络都有一个或两个全连接层,每层有128或256个节点。隐藏层之间的激活函数是'relu'函数。使用Adam优化器和1e-6的权重衰减来最大化ELBO。scMVAE模型使用pytorch软件包实现,其中GMM是使用Python scikit-learn软件包构建的。源代码位于GitHub存储库:https://github.com/cmzuo11/scMVAE。
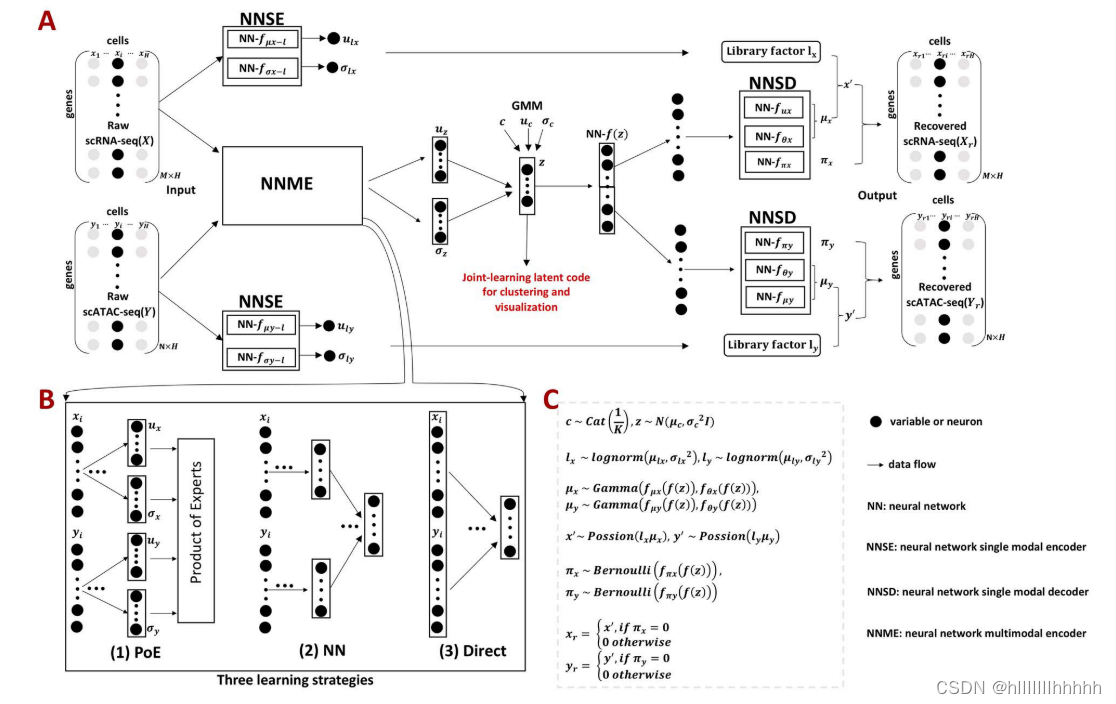
图1. scMVAE模型的概述,包括三种联合学习策略。
(A)scMVAE模型的总体框架。给定同一细胞i的scRNA-seq数据(具有M个变量的xi)和scATAC-seq数据(具有N个变量的yi)作为输入,scMVAE模型通过一个具有三种学习策略的多模态编码器学习了细胞的非线性联合嵌入(z),该嵌入可用于多种分析任务(例如细胞聚类和可视化),然后通过解码器对每个组学数据将其重构回原始维度作为输出。注意:两种组学数据的相同细胞顺序确保了一个细胞对应于低维空间中的一个点。
(B)三种学习策略的示意模型:
(i)‘PoE’框架用于通过每个组学数据的后验概率的乘积来估计联合后验(详见材料S1),
(ii)‘NN’用于通过使用神经网络来组合为每个层数据提取的特征来学习联合学习空间
(iii)‘Direct’策略通过直接使用两层数据的原始特征的串联作为输入一起学习。在这种学习条件下,神经网络:NN−fμy−l,NN−fσy−l,NN−fμy,NN−fθy,NN−fπy已从总网络中删除。
(C)scMVAE模型中每个变量所属的分布。每个组学数据都被建模为一个ZINB分布。有关每个变量的详细描述,请参见数据集和预处理。
这篇关于sc-MAVE的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!