本文主要是介绍化学分子Mol2文件格式与使用注意事项,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
| 欢迎浏览我的CSND博客! Blockbuater_drug …点击进入 |
|---|
前言
Mol2格式文件是一个ASCII 文件,由Tripos公司编制的用于表示化学分子的文件格式,在其药物设计软件套装SYBYL中使用。
Mol2格式文件被分子模拟的众多软件所支持,包括计算化学、分子对接,分子动力学软件,如Gaussian,VMD,UCSF DOCK,rDock,LeDock,MOE,Schrodinger,Openbabel,RDKit,Amber,Gromacs等。
Mol2格式文件可以通过关键词字段记录分子中常见的信息如原子坐标,原子类型,原子电荷,成键信息等,也可以记录晶胞常数、周期性边界条件信息和子结构信息等。
详细信息可参考: Mol2文件格式。
一、Mol2文件示例
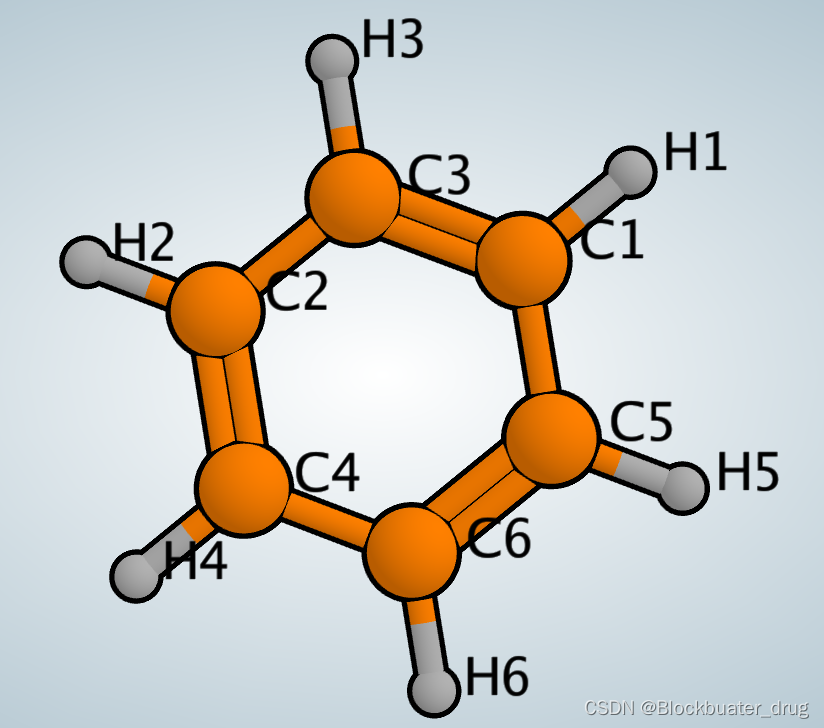
以下是苯环的Mol2格式文件:
# Name: benzene
# Creating user name: tom
# Creation time: Wed Dec 28 00:18:30 1988
# Modifying user name: tom
# Modification time: Wed Dec 28 00:18:30 1988@<TRIPOS>MOLECULE
benzene
12 12 1 0 0
SMALL
NO_CHARGES@<TRIPOS>ATOM1 C1 1.0787 0.8803 0.0002 C.ar 1 BENZENE 0.0000 2 H1 1.9151 1.5635 0.0008 H 1 BENZENE 0.0000 3 C2 -1.3021 0.4939 0.0002 C.ar 1 BENZENE 0.0000 4 H2 -2.3118 0.8772 0.0008 H 1 BENZENE 0.0000 5 C3 -0.2231 1.3734 -0.0002 C.ar 1 BENZENE 0.0000 6 H3 -0.3962 2.4394 -0.0008 H 1 BENZENE 0.0000 7 C4 -1.0787 -0.8803 -0.0002 C.ar 1 BENZENE 0.0000 8 H4 -1.9151 -1.5635 -0.0008 H 1 BENZENE 0.0000 9 C5 1.3021 -0.4939 -0.0002 C.ar 1 BENZENE 0.0000 10 H5 2.3118 -0.8772 -0.0008 H 1 BENZENE 0.0000 11 C6 0.2231 -1.3734 0.0002 C.ar 1 BENZENE 0.0000 12 H6 0.3962 -2.4394 0.0008 H 1 BENZENE 0.0000
@<TRIPOS>BOND1 1 2 1 2 1 5 ar 3 1 9 ar 4 3 4 1 5 3 5 ar 6 3 7 ar 7 5 6 1 8 7 8 1 9 7 11 ar 10 9 10 1 11 9 11 ar 12 11 12 1
@<TRIPOS>SUBSTRUCTURE1 BENZENE 11 PERM 0 **** **** 0 ROOT
二、 Mol2文件主要结构解释及注意事项
Mol2文件结构特点及使用注意事项:
- 允许注释和空行:前5行是注释,第6行是空行
- 接下来是4组关键字定义的结构信息,关键字以
@<TRIPOS>开头:@<TRIPOS>MOLECULE@<TRIPOS>ATOM@<TRIPOS>BOND@<TRIPOS>SUBSTRUCTURE,分别是分子信息,原子,键和子结构。具体解释如后文。 @<TRIPOS>MOLECULE@<TRIPOS>ATOM@<TRIPOS>BOND对于描述一个分子的结构信息是必须的。- Mol2文件格式相对自由,但在在不同关键字所在的组内有特定的编辑规则。
- 但不同软件对于原子类型描述可能有所不同,是某些软件读取出现错误的原因,如GaussView处理芳香系统,VMD里如果加载结构是采用xyz、gro、pdb等不含成键关系信息的坐标文件(虽然pdb有CONECT字段,但VMD不利用),保存出的mol2文件将没有BOND字段,缺失了成键信息,这种情况后续使用会有输入问题;如果在VMD同时加载相应的top或者prmtop等拓扑文件,再保存Mol2则可避免缺失BOND信息。
MOLECULE 字段解释
@<TRIPOS>MOLECULE 字段解释:
mol_name
num_atoms [num_bonds [num_subst [num_feat[num_sets]]]]
mol_type
charge_type
[status_bits
[mol_comment]]
每行信息解释:
第1行:mol_name体系的名字。OpenBabel把转换出mol2文件的源文件的名字作为体系的名字。
第2行:5个数字分别是体系中的原子数、化学键数、子结构数、特征数、set数。后三个可以省略。
子结构可以是体系中的一个部分,比如每个分子、每个残基、蛋白质的每条链等等都可以在@<TRIPOS>SUBSTRUCTURE字段里定义为一个子结构,不超过7个字符。
set是指基于体系中的一些原子/键/子结构根据特定规则和需要定义的集合,可以在@<TRIPOS>SET里具体定义,命名需要不重复。
第3行:mol_type体系的类型。可以为SMALL(小分子)、BIOPOLYMER(生物聚合物)、PROTEIN(蛋白)、NUCLEIC_ACID(核酸)、SACCHARIDE(糖)
第4行:原子电荷。没有计算就是NO_CHARGES,更具计算方法可以是GASTEIGER,MULLIKEN_CHARGES、MMFF94_CHARGES;其他情况USER_CHARGES。
方括号里面的信息项以及该项后面的项可以不提供。
ATOM 字段解释
@<TRIPOS>ATOM 字段,有10列:
atom_id atom_name x y z atom_type [subst_id [subst_name [charge [status_bit]]]]
每列信息解释:
(1)atom_id 原子序号(整数)
(2)atom_name 原子名(字符串)
(3)x坐标(埃)
(4)x坐标(埃)
(5)x坐标(埃)
(6)原子类型(字符串)
(7)原子所属的子结构序号(整数)
(8)原子所述的子结构名字(字符串)
(9)原子电荷(浮点数)
(10)状态位(status_bit):不可改动,由SYBYL根据原子产生,可省略:DSPMOD, TYPECOL, CAP, BACKBONE, DICT, ESSENTIAL, WATER 或者DIRECT.
方括号里面的信息可以不提供,不提供会被填充字符,保证至少9列。
第2列原子名称部分可以完全随意;但第6列原子类型Tripos有统一定义;
如下所示:

BOND 字段解释
@<TRIPOS>BOND 字段,有5列:
bond_id origin_atom_id target_atom_id bond_type [status_bits]
每列信息解释:
(1)bond_id键的序号(整数)
(2)origin_atom_id第1个原子的序号
(3)target_atom_id第2个原子的序号
(4)bond_type键的类型
(5)同ATOM可省略,用户不可修改。键的类型有以下这些:
• 1 = 单键
• 2 = 双键
• 3 = 三键
• am = 酰胺的N-C键(这种键有一定pi共轭作用,这是为什么mol2格式里特意用am来与单键区分)
• ar = 芳香环(aromatic)上的键,以下简称芳香键
• du = 虚键
• un = 未知/无法判断
• nc = 不相连(俩原子不成键就没必要在BOND字段出现,但可以靠nc强调某两个原子间就是没成键)
绝大多数程序产生的mol2文件里没有du、un、nc。
SUBSTRUCTURE字段解释
@<TRIPOS>SUBSTRUCTURE:字段解释:
subst_id subst_name root_atom [subst_type [dict_type [chain [sub_type [inter_bonds [status [comment]]]]]]]
方括号里面的信息可以不提供。subst_id子结构编号 (整数)
subst_name (字符串)
root_atom的编号 (整数)
subst_type:子结构类型(字符串)有:temp, perm, residue, group 或domain
Mol2文件所有关键词如下。进一步的信息可以参考 Mol2文件格式。

总结
本文提供了Mol2文件格式的解释,帮助初学者加深对结构文件的了解。
参考资料
- http://sobereva.com/655
- https://download.csdn.net/download/weixin_40192882/88872977?spm=1001.2014.3001.5503
| 欢迎浏览我的CSND博客! Blockbuater_drug …点击进入 |
|---|
这篇关于化学分子Mol2文件格式与使用注意事项的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!




