本文主要是介绍bedtools指南,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
文章目录
- 官方文档
- 下载安装
- 演示版的bed文件 (demo.bed)
- 我们的基因组文件(genome.txt)
- 两侧的运算
- 填充运算
- 下载测试数据
- 提取与genes.gff的间隔相对应的序列
- 获取测试数据
- 用这个间隔文件去分割bam文件
- 实战案例
- 获取数据
- bedtools intersect
- 从注释文件中,选取启动子
- 找到跟每个exon最近的启动子
- 以5Kb一个窗口把人类基因组以覆盖
官方文档
https://bedtools.readthedocs.io/en/latest/index.html
下载安装
cd ~/local/app/
curl -OL https://github.com/arq5x/bedtools2/releases/download/v2.22.0/bedtools-2.22.0.tar.gz
tar zxvf bedtools-2.22.0.tar.gz
cd bedtools2
make
ln -sf ~/local/app/bedtools2/bin/bedtools ~/bin/bedtools
演示版的bed文件 (demo.bed)
vim demo.bedKM034562 100 200 one 0 +
KM034562 400 500 two 0 -我们的基因组文件(genome.txt)
vim genome.txt
KM034562 18959两侧的运算
http://bedtools.readthedocs.io/en/latest/content/tools/flank.html
bedtools flank -i demo.gff -g genome.txt -b 10
KM034562 . . 91 100 0 + . .
KM034562 . . 201 210 0 + . .
KM034562 . . 391 400 0 - . .
KM034562 . . 501 510 0 - . .
bedtools flank -i demo.gff -g genome.txt -l 10 -r 0
KM034562 . . 91 100 0 + . .
KM034562 . . 391 400 0 - . .
bedtools flank -i demo.gff -g genome.txt -l 10 -r 0 -s > flank.gff
less flank.gff
KM034562 . . 91 100 0 + . .
KM034562 . . 501 510 0 - . .示意图:
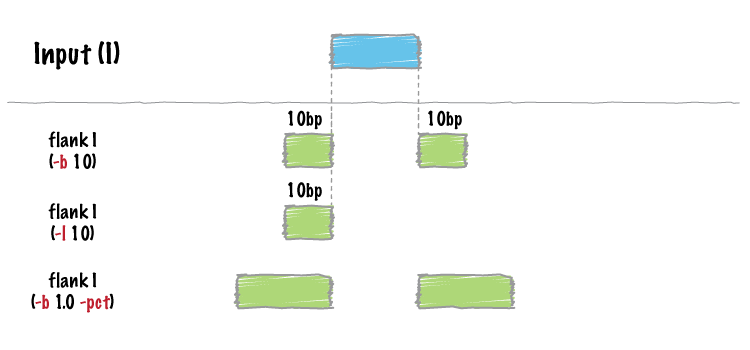
填充运算
http://bedtools.readthedocs.io/en/latest/content/tools/complement.html
bedtools complement -i demo.gff -g genome.txt > complement.gff
less complement.gff
KM034562 0 100
KM034562 200 400
KM034562 500 18959
示意图:
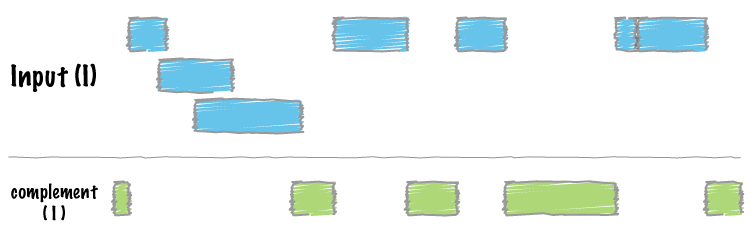
下载测试数据
通过Entrez Direct访问NCBI
efetch -id KM034562 -format gb -db nucleotide > KM034562.gb
数据格式转化
readseq -format=FASTA -o ~/data/852/KM034562.fa KM034562.gb
readseq -format=GFF -o KM034562.gff KM034562.gb
安装readseq
创建 ~/bin 目录
mkdir -p ~/bin
将~/bin 文件夹加到PATH
echo 'export PATH=~/bin:$PATH' >> ~/.bashrc
source ~/.bashrc-------------------------------------
mkdir ~/local/app/readseq
cd ~/local/app/readseq
curl -OL http://iubio.bio.indiana.edu/soft/molbio/readseq/java/readseq.jar-----------------------------------------
echo '#!/bin/bash' > ~/bin/readseq
echo 'java -jar ~/local/app/readseq/readseq.jar $@' >> ~/bin/readseq
chmod +x ~/bin/readseq从整个文件中提取基因
cat KM034562.gff | bioawk -c gff ' $feature=="gene" { print $0 } ' > genes.gff
less genes.gff
KM034562 - gene 56 3026 . + . gene "NP"
KM034562 - gene 3032 4407 . + . gene "VP35"
KM034562 - gene 4390 5894 . + . gene "VP40"
KM034562 - gene 5900 8305 . + . gene "GP"
KM034562 - gene 8288 9740 . + . gene "VP30"
KM034562 - gene 9885 11518 . + . gene "VP24" ; note "putative"
KM034562 - gene 11501 18282 . + . gene "L"
提取与genes.gff的间隔相对应的序列
http://bedtools.readthedocs.io/en/latest/content/tools/getfasta.html
bedtools getfasta -fi ~/data/852/KM034562.fa -bed genes.gff -fo genes.fa
head genes.fa
示意图:

获取测试数据
mkdir -p ~/local/app
cd ~/local/app
wget https://sourceforge.net/projects/bowtie-bio/files/bowtie2/2.3.4.3/bowtie2-2.3.4.3-linux-x86_64.zipunzip bowtie2-2.3.4.3-linux-x86_64.zip
ln -s ~/local/app/bowtie2-2.3.4.3-linux-x86_64/bowtie2 ~/bincd ~/local/app/bowtie2-2.3.4.3-linux-x86_64/example/reads/bowtie2 -x ../index/lambda_virus -1 reads_1.fq -2 reads_2.fq 2>alignment.log |samtools view -bS >tmp.bamsamtools sort tmp.bam > tmp.sorted.bamsamtools index tmp.sorted.bam
用这个间隔文件去分割bam文件
http://bedtools.readthedocs.io/en/latest/content/tools/intersect.html
创建一个间隔文件
vim region.bed
gi|9626243|ref|NC_001416.1| 1000 2000[sunchengquan 11:48:40 ~/local/app/bowtie2-2.3.4.3-linux-x86_64/example/reads]
$ bedtools intersect -a tmp.sorted.bam -b region.bed > region.bam
[sunchengquan 11:50:34 ~/local/app/bowtie2-2.3.4.3-linux-x86_64/example/reads]
$ wc -l tmp.sorted.bam
10124 tmp.sorted.bam
[sunchengquan 11:50:47 ~/local/app/bowtie2-2.3.4.3-linux-x86_64/example/reads]
$ wc -l region.bam
220 region.bam
[sunchengquan 11:52:59 ~/local/app/bowtie2-2.3.4.3-linux-x86_64/example/reads]
$ samtools view -h region.bam |awk '{print $4}'|tail -1
2000
示意图:
实战案例
http://quinlanlab.org/tutorials/bedtools/bedtools.html
获取数据
mkdir ~/edu/lec28
cd ~/edu/lec28
curl -O https://s3.amazonaws.com/bedtools-tutorials/web/maurano.dnaseI.tgz
curl -O https://s3.amazonaws.com/bedtools-tutorials/web/cpg.bed
curl -O https://s3.amazonaws.com/bedtools-tutorials/web/exons.bed
curl -O https://s3.amazonaws.com/bedtools-tutorials/web/gwas.bed
curl -O https://s3.amazonaws.com/bedtools-tutorials/web/genome.txt
curl -O https://s3.amazonaws.com/bedtools-tutorials/web/hesc.chromHmm.bed
tar -zxvf maurano.dnaseI.tgz
rm maurano.dnaseI.tgz
这些文件内容:
胎儿的组织样品,包括脑、心脏、肠道、肾脏、肺、肌肉、皮肤以及胃
cpg.bed
人类基因组中的CpG岛
exons.bed
RefSeq exons from human genes
gwas.bed
human disease-associated SNPs that were identified in genome-wide association studies (GWAS)
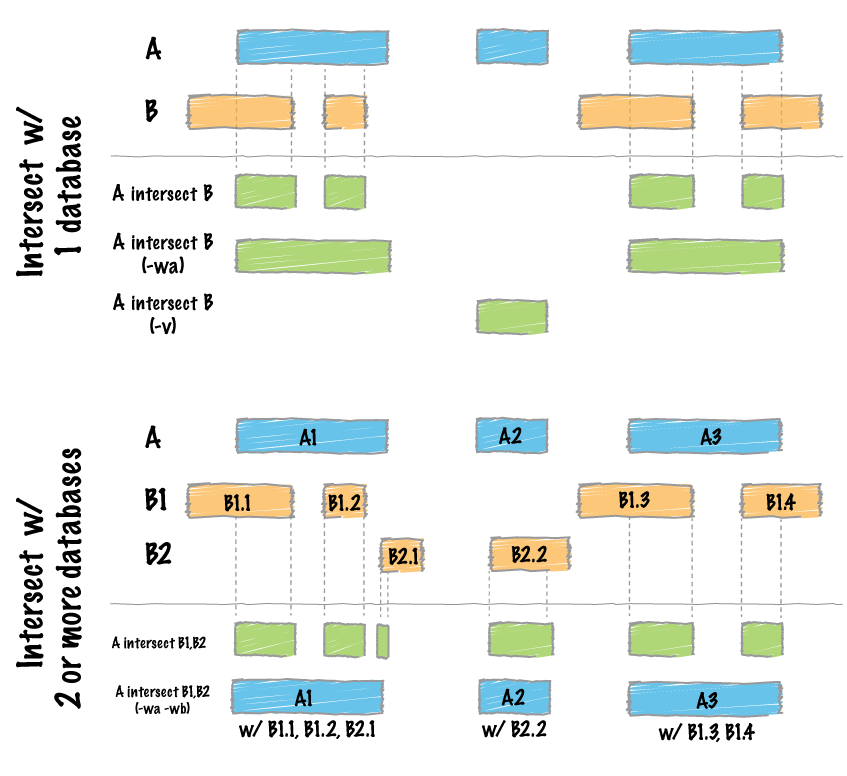
bedtools intersect
#找到A和B文件中重叠的部分
bedtools intersect -a cpg.bed -b exons.bed | head -5
chr1 29320 29370 CpG:_116
chr1 135124 135563 CpG:_30
chr1 327790 328229 CpG:_29
chr1 327790 328229 CpG:_29
chr1 327790 328229 CpG:_29#-wa:A和B重叠的区间再加上a的剩余部分
#-wb:A和B重叠的区间再加上b的剩余部分
bedtools intersect -a cpg.bed -b exons.bed -wa -wb | head -5
chr1 28735 29810 CpG:_116 chr1 29320 29370 NR_024540_exon_10_0_chr1_29321_r 0 -
chr1 135124 135563 CpG:_30 chr1 134772 139696 NR_039983_exon_0_0_chr1_134773_r 0 -
chr1 327790 328229 CpG:_29 chr1 324438 328581 NR_028322_exon_2_0_chr1_324439_f 0 +
chr1 327790 328229 CpG:_29 chr1 324438 328581 NR_028325_exon_2_0_chr1_324439_f 0 +
chr1 327790 328229 CpG:_29 chr1 327035 328581 NR_028327_exon_3_0_chr1_327036_f 0 +#-wo Write the original A and B entries plus the number of base pairs of overlap between the two features. Only A features with overlap are reported
bedtools intersect -a cpg.bed -b exons.bed -wo | head -5
chr1 28735 29810 CpG:_116 chr1 29320 29370 NR_024540_exon_10_0_chr1_29321_r 0 - 50
chr1 135124 135563 CpG:_30 chr1 134772 139696 NR_039983_exon_0_0_chr1_134773_r 0 - 439
chr1 327790 328229 CpG:_29 chr1 324438 328581 NR_028322_exon_2_0_chr1_324439_f 0 + 439
chr1 327790 328229 CpG:_29 chr1 324438 328581 NR_028325_exon_2_0_chr1_324439_f 0 + 439
chr1 327790 328229 CpG:_29 chr1 327035 328581 NR_028327_exon_3_0_chr1_327036_f 0 + 439 #-c For each entry in A, report the number of hits in B while restricting to -f. Reports 0 for A entries that have no overlap with B
bedtools intersect -a cpg.bed -b exons.bed -c | head
chr1 28735 29810 CpG:_116 1
chr1 135124 135563 CpG:_30 1
chr1 327790 328229 CpG:_29 3
chr1 437151 438164 CpG:_84 0
chr1 449273 450544 CpG:_99 0
chr1 533219 534114 CpG:_94 0
chr1 544738 546649 CpG:_171 0
chr1 713984 714547 CpG:_60 1
chr1 762416 763445 CpG:_115 10
chr1 788863 789211 CpG:_28 9# 找到覆盖了最多外显子的CPG岛
bedtools intersect -a cpg.bed -b exons.bed -c | sort -k5,5nr | head -2
chrY 15591259 15591720 CpG:_33 77
chrUn_gl000228 70214 114054 CpG:_3259 72bedtools intersect -a cpg.bed -b exons.bed -c | sort -k1,1 -k2,2nr | head -2
chr1 249200252 249200721 CpG:_58 2
chr1 249167408 249168010 CpG:_48 0#找到A文件中没有重叠B的部分 Only report those entries in A that have no overlap in B
bedtools intersect -a cpg.bed -b exons.bed -v | head
chr1 437151 438164 CpG:_84
chr1 449273 450544 CpG:_99
chr1 533219 534114 CpG:_94
chr1 544738 546649 CpG:_171
chr1 801975 802338 CpG:_24
chr1 805198 805628 CpG:_50
chr1 839694 840619 CpG:_83
chr1 844299 845883 CpG:_153
chr1 912869 913153 CpG:_28
chr1 919726 919927 CpG:_15
从注释文件中,选取启动子
cat hesc.chromHmm.bed | grep Promoter > promoters.bed
cat promoters.bed |head -3
chr1 27737 28537 2_Weak_Promoter
chr1 28537 30137 1_Active_Promoter
chr1 30137 30337 2_Weak_Promoter
找到跟每个exon最近的启动子
多的一列数值是-a 和 -b 两者最近的距离
bedtools closest -a exons.bed -b promoters.bed -d | head -2
chr1 11873 12227 NR_046018_exon_0_0_chr1_11874_f 0 + chr1 27737 28537 2_Weak_Promoter 15511
chr1 12612 12721 NR_046018_exon_1_0_chr1_12613_f 0 + chr1 27737 28537 2_Weak_Promoter 15017
- -d In addition to the closest feature in B, report its distance to A as an extra column. The reported distance for overlapping features will be 0
示意图:

以5Kb一个窗口把人类基因组以覆盖
bedtools makewindows -g genome.txt -w 50000 > windows.bed
cat windows.bed |head -3
chr1 0 50000
chr1 50000 100000
chr1 100000 150000
bedtools makewindows -g genome.txt -w 100000 > windows0.bed
cat windows0.bed |head -3
chr1 0 100000
chr1 100000 200000
chr1 200000 300000
这篇关于bedtools指南的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!





