本文主要是介绍【哈佛大学生物信息学与计算生物学】视频笔记,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
目录
- 生物信息学研究浪潮
- Bioinfo vs Computational Biology
- Levels of Bioinfo / Comp Bio
- 生物信息学相关学科 & 技术
- FASTQ File
- fastqc对原始测序reads质控
- Per Base Sequence Quality
- Per Sequence Quality Distribution
- Nucleotide Content Per Position
- Per Sequence GC Content
- 局部比对算法
- RPKM、FPKM 、TPM
- RNA-Seq Read Distribution
- independent filtering
- FDR
- 聚类
生物信息学研究浪潮
- 研究蛋白序列和结构
- 基因表达(微阵列技术)
- DNA测序
基因组测序成本下降非常快,可以用于个性化诊断,eg:肿瘤序列-靶向疗法
Bioinfo vs Computational Biology
-
第一代测序:桑格测序 Sanger sequencing(需要扩增dna,并且每次只扩增一个分子,所以需要扩增很多次同一单链DNA的多个拷贝)
-
第二代测序:大规模平行测序 Illumina Sequencing Cluster Generation(不需要将不同DNA分装到不同的管子当中进行扩增,可以将很多很多个分子倒在流动池上进行原位扩增,边合成边测序)如果需要产生很多很多的片段,需要了解定量水平,第二代测序更符合要求。

-
第三代测序:真正的单分子测序,甚至再也不需要扩增dna。一般来讲,会使用聚合酶或微孔进行测序,然后根据acgt当中哪个核苷酸被结合或通过微孔而发出不同的信号。

Levels of Bioinfo / Comp Bio

学无止境!!!
生物信息学相关学科 & 技术

FASTQ File
- 第一行 序列id
- 第二行 真实序列
- 第三行 序列名称,与第一行不同的前缀分别是@和+,@所在行代表路序列的名称(以及可选的描述内容)
+后面可以是序列名,也可以为空 - 第四行 序列值,由33个不同的ASCII字符表示
ASCII数值所大致代表的Phred质量的计算公式是-10*log10 Pr(碱基对出错的概率)
通常来说,质量值的数字越大,质量就越高,出错的概率越小。

fastqc对原始测序reads质控
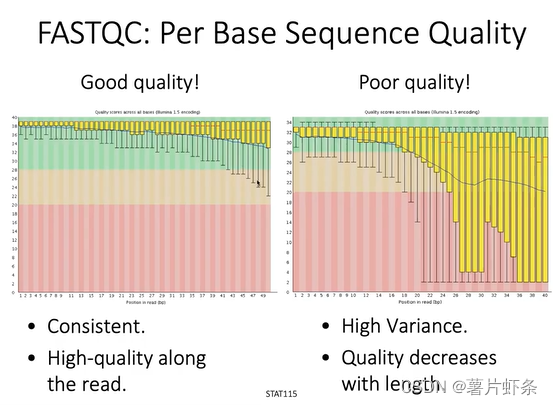
Per Base Sequence Quality
高质量序列的碱基其实是高质量并连续出现的。
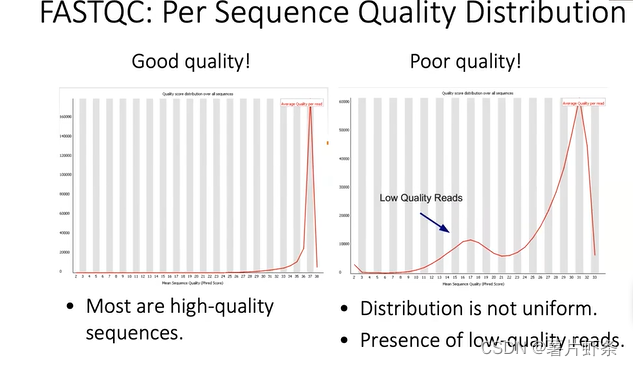
Per Sequence Quality Distribution
碱基对应质量值的分布
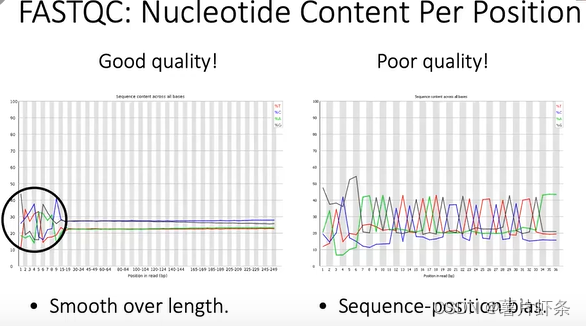
Nucleotide Content Per Position
在全基因组水平进行测序,每一种类型的碱基其占比应在25%左右。
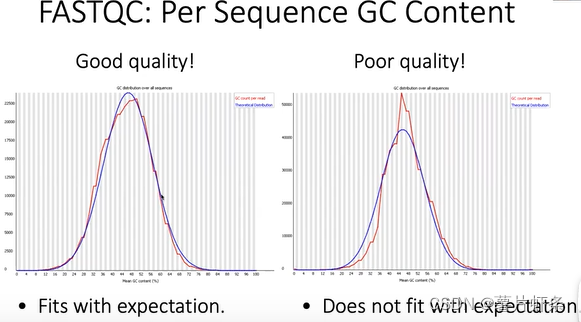
Per Sequence GC Content
将实际的GC content与期望的GC content进行对比,右图意味着测序也许出现了问题。
局部比对算法
局部比对是一种序列比对技术,在该技术中,我们对比两个序列之间具有较高相似性的区域,即具有最高匹配密度的序列的延伸,这适用于更多部分相似且在序列之间具有保守区域的转向序列。
smith waterman算法是最常见的局部比对,属于动态规划,讲一个问题分解为更小更简单的子问题。
全局比对试图将整个序列彼此比对,而局部比对仅比对这些具有最高相似性。全局比对比对来自查询序列和目标序列的所有字母,而局部比对将目标序列的子串与查询序列的子串对齐。
因此,全局比对更适合密切相关的序列,而局部对比更适合发散或远相关的序列。
序列最常见的全局比对算法是needleman-wunsch,而局部比对是smith-waterman。
RPKM、FPKM 、TPM
三者都是衡量基因相对表达量
RPKM(Reads Per Kilobase per Million)
FPKM(Fragments Per Kilobase per Million)
TPM:Transcripts Per Kilobase per Million mapped reads (每千个碱基的转录每百万映射读取的Transcripts)
RPKM和FPKM的计算方法:第一步先将测序深度标准化,第二步是基因长度的标准化,即将第一步的read per million直接除以基因长度即可。
TPM的不同在于它的处理顺序是不同的。即先考虑基因长度,再考虑测序深度。

RNA-Seq Read Distribution
在芯片时代,RNA-Seq测序的reads分布,一般被认为是正态分布。
RNA-Seq测序得到的reads分布,一般符合泊松分布。
independent filtering
检测差异基因表达的方法是进行对每一个基因进行统计检验。
所谓的independent filtering,意思是在进行统计检验之前,筛掉那些不能或是很可能不能通过显著性检验的探针。independent filtering就是为了降低假设检验的假阴性。
FDR

与GO富集分析的差异在于GSEA分析不需要指定阈值(p值或FDR)来筛选差异基因。
聚类
heatmap(热力图),通过颜色的深浅程度来判断不同类别间的差异,呈现不同特征间的聚类关系,通常用做聚类分析(如层次聚类,kmeans聚类等)。
Hierarchical Clustering(层次聚类)
自上而下:分裂法,初始时将所有的样本归为一个类簇,然后依据某种准则进行逐渐的分裂,直到达到某种条件或者达到设定的分类数目。
自下而上:凝聚法,初始时将每个样本点当做一个类簇,所以原始类簇的大小等于样本点的个数,然后依据某种准则合并这些初始的类簇,直到达到某种条件或者达到设定的分类数目。
K-means Clustering(k均值聚类),基于样本集合划分的聚类算法。
Consensus Clustering(一致性聚类),被广泛用于基于亚群鉴定和癌症分型等研究方向,采用重抽样方法打乱原始数据集,对每一次聚类的样本进行聚类分析,最后再综合评估多次聚类分析的结果给出一致性。


这篇关于【哈佛大学生物信息学与计算生物学】视频笔记的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!





