本文主要是介绍挑选富集分析结果 enrichments,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
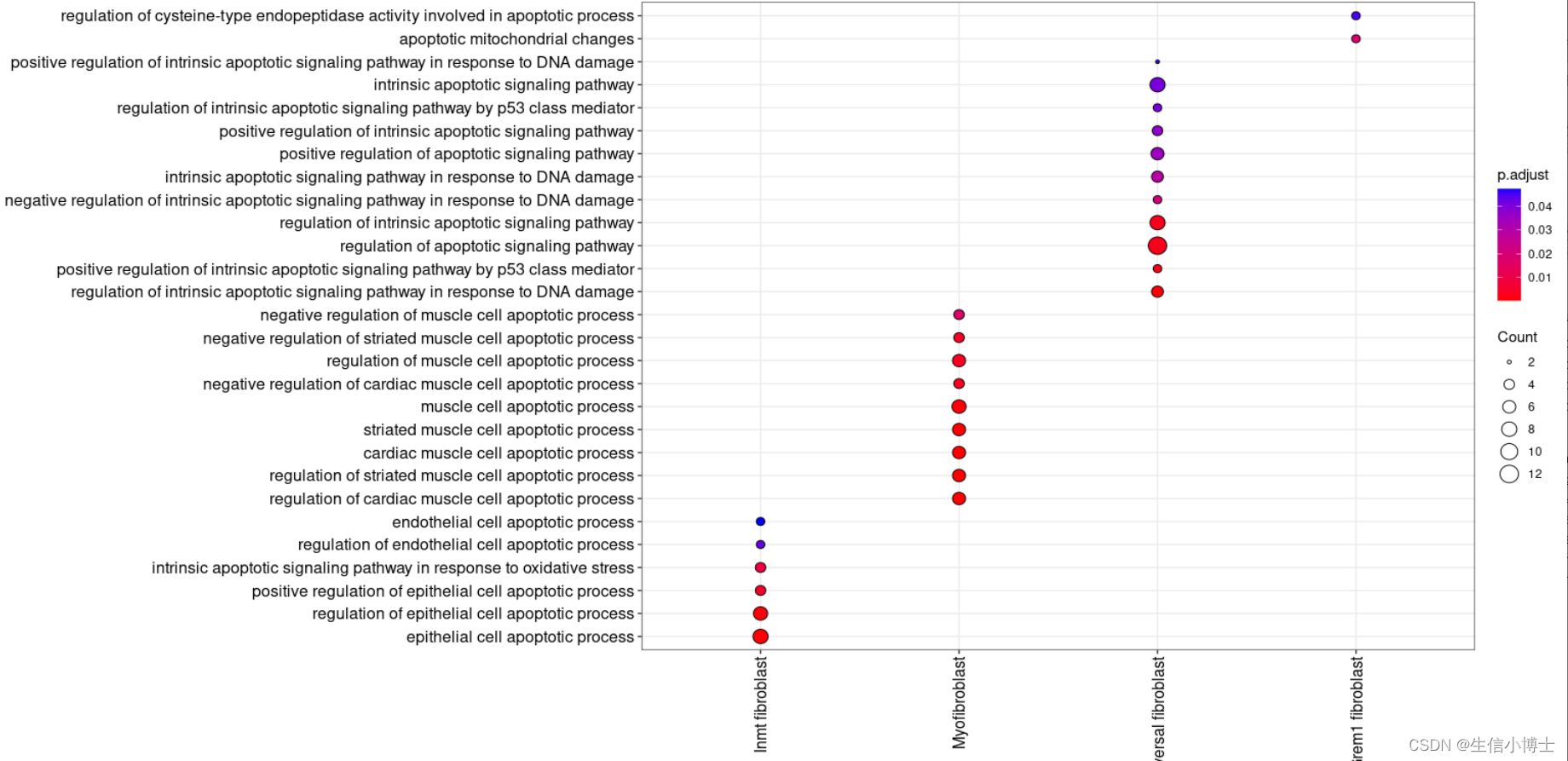
#2.2挑选term---selected_clusterenrich=enrichmets[grepl(pattern = "cilium|matrix|excular|BMP|inflamm|development|muscle|vaso|pulmonary|alveoli",x = enrichmets$Description),]head(selected_clusterenrich) distinct(selected_clusterenrich)# remove duplicate rows based on Description 并且保留其他所有变量
distinct_df <- distinct(enrichmets, Description,.keep_all = TRUE)library(ggplot2)
ggplot( distinct_df %>%dplyr::filter(stringr::str_detect(pattern = "cilium|matrix|excular|BMP|inflamm|development|muscle",Description)) %>%group_by(Description) %>%add_count() %>%dplyr::arrange(dplyr::desc(n),dplyr::desc(Description)) %>%mutate(Description =forcats:: fct_inorder(Description)), #fibri|matrix|collaaes(Cluster, Description)) +geom_point(aes(fill=p.adjust, size=Count), shape=21)+theme_bw()+theme(axis.text.x=element_text(angle=90,hjust = 1,vjust=0.5),axis.text.y=element_text(size = 12),axis.text = element_text(color = 'black', size = 12))+scale_fill_gradient(low="red",high="blue")+labs(x=NULL,y=NULL)
# coord_flip()head(enrichmets)ggplot( distinct(enrichmets,Description,.keep_all=TRUE) %>% # dplyr::mutate(Cluster = factor(Cluster, levels = unique(.$Cluster))) %>%dplyr::mutate(Description = factor(Description, levels = unique(.$Description))) %>%# dplyr::group_by(Cluster) %>%dplyr::filter(stringr::str_detect(pattern = "cilium organization|motile cilium|cilium movemen|cilium assembly|cell-matrix adhesion|extracellular matrix organization|regulation of acute inflammatory response to antigenic stimulus|collagen-containing extracellular matrix|negative regulation of BMP signaling pathway|extracellular matrix structural constituent|extracellular matrix binding|fibroblast proliferation|collagen biosynthetic process|collagen trimer|fibrillar collagen trimer|inflammatory response to antigenic stimulus|chemokine activity|chemokine production|cell chemotaxis|chemoattractant activity|NLRP3 inflammasome complex assembly|inflammatory response to wounding|Wnt signaling pathway|response to oxidative stress|regulation of vascular associated smooth muscle cell proliferation|venous blood vessel development|regulation of developmental growth|lung alveolus development|myofibril assembly|blood vessel diameter maintenance|gas transport|cell maturation|regionalization|oxygen carrier activity|oxygen binding|vascular associated smooth muscle cell proliferation",Description)) %>%# group_by(Description) %>%add_count() %>%dplyr::arrange(dplyr::desc(n),dplyr::desc(Description)) %>%mutate(Description =forcats:: fct_inorder(Description)), #fibri|matrix|collaaes(Cluster, y = Description)) + #stringr:: str_wrapgeom_point(aes(fill=p.adjust, size=Count), shape=21)+theme_bw()+theme(axis.text.x=element_text(angle=90,hjust = 1,vjust=0.5),axis.text.y=element_text(size = 12),axis.text = element_text(color = 'black', size = 12))+scale_fill_gradient(low="red",high="blue")+labs(x=NULL,y=NULL)
# coord_flip()
print(getwd())p=ggplot( distinct(enrichmets,Description,.keep_all=TRUE) %>%dplyr::mutate(Description = factor(Description, levels = unique(.$Description))) %>% #调整terms显示顺序dplyr::filter(stringr::str_detect(pattern = "cilium organization|motile cilium|cilium movemen|cilium assembly|cell-matrix adhesion|extracellular matrix organization|regulation of acute inflammatory response to antigenic stimulus|collagen-containing extracellular matrix|negative regulation of BMP signaling pathway|extracellular matrix structural constituent|extracellular matrix binding|fibroblast proliferation|collagen biosynthetic process|collagen trimer|fibrillar collagen trimer|inflammatory response to antigenic stimulus|chemokine activity|chemokine production|cell chemotaxis|chemoattractant activity|NLRP3 inflammasome complex assembly|inflammatory response to wounding|Wnt signaling pathway|response to oxidative stress|regulation of vascular associated smooth muscle cell proliferation|venous blood vessel development|regulation of developmental growth|lung alveolus development|myofibril assembly|blood vessel diameter maintenance|gas transport|cell maturation|regionalization|oxygen carrier activity|oxygen binding|vascular associated smooth muscle cell proliferation",Description)) %>%group_by(Description) %>%add_count() %>%dplyr::arrange(dplyr::desc(n),dplyr::desc(Description)) %>%mutate(Description =forcats:: fct_inorder(Description)), #fibri|matrix|collaaes(Cluster, y = Description)) + #stringr:: str_wrap#scale_y_discrete(labels = function(x) stringr::str_wrap(x, width = 60)) + #调整terms长度geom_point(aes(fill=p.adjust, size=Count), shape=21)+theme_bw()+theme(axis.text.x=element_text(angle=90,hjust = 1,vjust=0.5),axis.text.y=element_text(size = 12),axis.text = element_text(color = 'black', size = 12))+scale_fill_gradient(low="red",high="blue")+labs(x=NULL,y=NULL)
# coord_flip()
print(getwd())ggsave(filename ="~/silicosis/spatial/sp_cluster_rigions_after_harmony/enrichents12.pdf",plot = p,width = 10,height = 12,limitsize = FALSE)######展示term内所有基因,用热图展示-------#提取画图的数据p$data#提取图形中的所有基因-----
mygenes= p$data $geneID %>% stringr::str_split(.,"/",simplify = TRUE) %>%as.vector() %>%unique()
frame_for_genes=p$data %>%as.data.frame() %>% dplyr::group_by(Cluster) #后面使用split的话,必须按照分组排序
head(frame_for_genes)my_genelist= split(frame_for_genes, frame_for_genes$Cluster, drop = TRUE) %>% #注意drop参数的理解lapply(function(x) select(x, geneID));my_genelistmy_genelist= split(frame_for_genes, frame_for_genes$Cluster, drop = TRUE) %>% #注意drop参数的理解lapply(function(x) x$geneID);my_genelistmygenes=my_genelist %>% lapply( function(x) {stringr::str_split(x,"/",simplify = TRUE) %>%as.vector() %>%unique()} )#准备画热图,加载seurat对象
load("/home/data/t040413/silicosis/spatial_transcriptomics/silicosis_ST_harmony_SCT_r0.5.rds")
{dim(d.all)DefaultAssay(d.all)="Spatial"#visium_slides=SplitObject(object = d.all,split.by = "stim")names(d.all);dim(d.all)d.all@meta.data %>%head()head(colnames(d.all))#1 给d.all 添加meta信息------adata_obs=read.csv("~/silicosis/spatial/adata_obs.csv")head(adata_obs)mymeta= paste0(d.all@meta.data$orig.ident,"_",colnames(d.all)) %>% gsub("-.*","",.) # %>% head()head(mymeta)tail(mymeta)#掉-及其之后内容adata_obs$col= adata_obs$spot_id %>% gsub("-.*","",.) # %>% head()head(adata_obs)rownames(adata_obs)=adata_obs$coladata_obs=adata_obs[mymeta,]head(adata_obs)identical(mymeta,adata_obs$col)d.all=AddMetaData(d.all,metadata = adata_obs)head(d.all@meta.data)}##构建画热图对象---
Idents(d.all)=d.all$clusters
a=AverageExpression(d.all,return.seurat = TRUE)
a$orig.ident=rownames(a@meta.data)
head(a@meta.data)
head(markers)rownames(a) %>%head()
head(mygenes)
table(mygenes %in% rownames(a))
DoHeatmap(a,draw.lines = FALSE, slot = 'scale.data', group.by = 'orig.ident',features = mygenes ) + ggplot2:: scale_color_discrete(name = "Identity", labels = unique(a$orig.ident) %>%sort() )##doheatmap做出来的图不好调整,换成heatmap自己调整p=DoHeatmap(a,draw.lines = FALSE, slot = 'scale.data', group.by = 'orig.ident',features = mygenes ) + ggplot2:: scale_color_discrete( labels = unique(a$orig.ident) %>%sort() ) #name = "Identity",p$data %>%head()##########这种方式容易出现bug,不建议------
if (F) {wide_data <- p$data %>% .[,-4] %>%tidyr:: pivot_wider(names_from = Cell, values_from = Expression)print(wide_data) mydata= wide_data %>%dplyr:: select(-Feature) %>%as.matrix()head(mydata)rownames(mydata)=wide_data$Featuremydata=mydata[,c("Bronchial zone", "Fibrogenic zone", "Interstitial zone", "Inflammatory zone","Vascular zone" )]p2=pheatmap:: pheatmap(mydata, fontsize_row = 2, clustering_method = "ward.D2",# annotation_col = wide_data$Feature,annotation_colors = c("Interstitial zone" = "red", "Bronchial zone" = "blue", "Fibrogenic zone" = "green", "Vascular zone" = "purple") ,cluster_cols = FALSE,column_order = c("Inflammatory zone", "Vascular zone" ,"Bronchial zone", "Fibrogenic zone" ))getwd()ggplot2::ggsave(filename = "~/silicosis/spatial/sp_cluster_rigions_after_harmony/heatmap_usingpheatmap.pdf",width = 8,height = 10,limitsize = FALSE,plot = p2)}##########建议如下方式画热图------
a$orig.ident=a@meta.data %>%rownames()
a@meta.data %>%head()
Idents(a)=a$orig.identa@assays$Spatial@scale.data %>%head()mydata=a@assays$Spatial@scale.data
mydata=mydata[rownames(mydata) %in% (mygenes %>%unlist() %>%unique()) ,]
mydata= mydata[,c( "Fibrogenic zone", "Inflammatory zone", "Bronchial zone","Interstitial zone","Vascular zone" )]
head(mydata)
p3=pheatmap:: pheatmap(mydata, fontsize_row = 2, clustering_method = "ward.D2",# annotation_col = wide_data$Feature,annotation_colors = c("Interstitial zone" = "red", "Bronchial zone" = "blue", "Fibrogenic zone" = "green", "Vascular zone" = "purple") ,cluster_cols = FALSE,column_order = c("Inflammatory zone", "Vascular zone" ,"Bronchial zone", "Fibrogenic zone" )
)getwd()
ggplot2::ggsave(filename = "~/silicosis/spatial/sp_cluster_rigions_after_harmony/heatmap_usingpheatmap2.pdf",width = 8,height = 10,limitsize = FALSE,plot = p3)#########单独画出炎症区和纤维化区---------
a@assays$Spatial@scale.data %>%head()mydata=a@assays$Spatial@scale.data
mygenes2= my_genelist[c('Inflammatory zone','Fibrogenic zone')] %>% unlist() %>% stringr::str_split("/",simplify = TRUE) mydata2=mydata[rownames(mydata) %in% ( mygenes2 %>%unlist() %>%unique()) ,]
mydata2= mydata2[,c( "Fibrogenic zone", "Inflammatory zone" )]
head(mydata2)p3=pheatmap:: pheatmap(mydata2, fontsize_row = 5, #scale = 'row',clustering_method = "ward.D2",# annotation_col = wide_data$Feature,annotation_colors = c("Interstitial zone" = "red", "Bronchial zone" = "blue", "Fibrogenic zone" = "green", "Vascular zone" = "purple") ,cluster_cols = FALSE,column_order = c("Inflammatory zone", "Vascular zone" ,"Bronchial zone", "Fibrogenic zone" )
)getwd()
ggplot2::ggsave(filename = "~/silicosis/spatial/sp_cluster_rigions_after_harmony/heatmap_usingpheatmap3.pdf",width = 4,height = 8,limitsize = FALSE,plot = p3)这篇关于挑选富集分析结果 enrichments的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!



