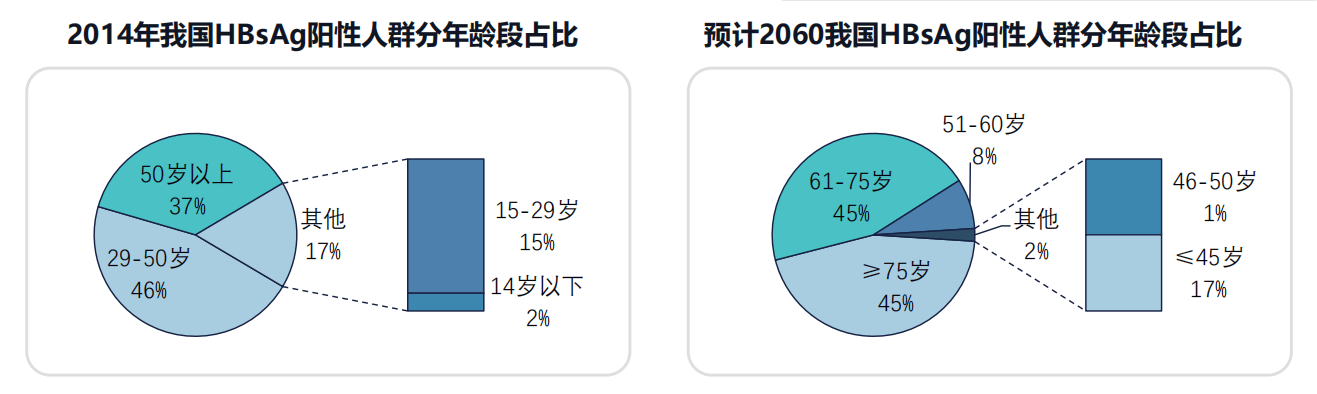
本文主要是介绍H3.3K27M弥漫性中线胶质瘤的反义寡核苷酸治疗,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
今天给同学们分享一篇实验文章“Antisense oligonucleotide therapy for H3.3K27M diffuse midline glioma”,这篇文章发表在Sci Transl Med期刊上,影响因子为17.1。

结果解读:
CRISPR-Cas9消耗H3.3K27M恢复了H3K27三甲基化,并延缓了患者来源的神经球和原位异种移植物的生长
作为对本研究中使用的模型中完全基因敲除H3-3A的生物学后果的控制,作者首先使用CRISPR-Cas9和sgRNA靶向突变和野生型等位基因,但不影响编码相同蛋白质的H3-3B,在两个DIPG患者细胞系SU-DIPG-XIII和XVII中(9)。K27M突变对H3K27me3总量的显性负效应已有充分的文献证明(1-4)。在携带H3-3A杂合突变的两个患者细胞系中,H3.3K27M敲除恢复了抑制性的H3K27me3三甲基化标记,并减少了通过免疫印迹检测到的允许性的H3K27ac乙酰化标记。转导gRNA抗性H3-3A cDNA降低了恢复的H3K27me3标记,与靶向效应一致。敲除恢复了H2K27me3三甲基化并减少了细胞增殖,通过免疫荧光、EdU染色以及软琼脂集落形成进行检测。为了确定H3.3K27M在体内需要用于肿瘤维持,作者在免疫缺陷的非肥胖糖尿病、严重联合免疫缺陷病、伽玛(NSG)小鼠的出生后第3天(P3)进行了患者细胞或H3-3A K27M 基因敲除细胞的原位移植(9)。作者选择了SU-DIPG-XIII系列进行此实验,因为其较慢的增殖速率扩大了作者治疗的窗口期。移植的敲除细胞显示出延迟的肿瘤潜伏期,导致显著增加的生存率(P = 0.039)。与正常相邻组织相比,H3.3K27M异种移植瘤在组织学上与患者肿瘤相似,H3K27me3标记全局减少。这种效应在敲除瘤中得到缓解,并与成熟神经元标记物神经核抗原(NeuN)和星形胶质纤维酸性蛋白(GFAP)的表达升高相关,通过免疫荧光检测到,这表明敲除细胞在体内增殖较少且更倾向于分化的谱系。这些结果证实并扩展了先前的研究(5, 6),并强调了突变H3的潜力。3K27M作为治疗靶点。
Gapmer ASO筛选以减少突变的H3-3A mRNA和H3.3K27M蛋白质
为了在DIPG患者细胞中药理学地靶向H3-3A mRNA,作者探索了ASOs的潜力。作者设计并测试了带有2'-O-甲氧基乙基(MOE)翼和磷酸硫酸酯(PS)骨架的gapmer ASOs。这些gapmer ASOs具有类似DNA的中央区域,通过内源性RNase H引导对互补mRNA或前mRNA靶点的剪切,并具有化学修饰的翼,促进更紧密的RNA结合,增强稳定性和改善细胞摄取(12)。首先,为了尝试实现靶向等位基因特异性沉默,作者设计了10个交集的20-mer PS-MOE-ASOs(#1-10),跨越H3-3A外显子2的突变位点和相邻核苷酸,突变位点位于ASO间隙上。这些序列与H3-3A K27M 等位基因完全互补,并与H3-3A WT 等位基因有一个错配。其次,为了实现靶向基因特异性沉默-考虑到野生型H3.3蛋白仍然由H3-3B基因表达-作者设计了另外五个交集的20-mer PS-MOE gapmer ASOs(#11-15),以靶向外显子2突变位点上游或下游的外显子区域。这些ASO仅设计用于靶向H3-3A转录本,而不是H3-3B转录本,或者编码H3.1或H3.2经典组蛋白蛋白质的其他基因转录本。此外,作者通过克隆包含外显子1至3、完整内含子1和2的基因组片段,生成了一个野生型H3-3A迷你基因和一个带有A到T突变的突变型H3-3A迷你基因。
为了测试这些ASOs是否介导RNase-H1对H3-3A K27M mRNA的剪切,或者对突变型和野生型H3-3A都起作用,作者将单个ASOs(100 nM)与H3-3A WT 或H3-3A K27M 小基因共转染到HeLa细胞中。作者还通过自由摄取(4 μM)将这些ASOs传递到患者来源的神经球培养中。后一种方法依赖于受体介导的内吞作用,并且需要更高浓度的ASO(12)。作者的初步小基因筛选确定了三个连续的具有等位基因特异设计的ASOs(ASO4、5和6),以及两个连续的具有基因特异设计的ASOs(ASO12和13),它们实现了H3-3A的强效沉默(图1A)。所有等位基因特异的ASOs在患者来源的细胞中对突变型等位基因的沉默效果更强,而在这个浓度下,ASO4、5和6在小基因环境中都能强效沉默两个等位基因。同样,通过自由摄取传递到神经球培养中的ASOs将内源性突变等位基因的表达降低了50-70%,而野生型等位基因的表达降低程度较小(30-40%)(图1B)。出乎意料的是,一种基因特异性的ASO,ASO15,也促进了等位基因特异性的沉默,这表明了在其靶区域内结合的可能的调控性RNA结合蛋白的立体阻断(图1B)。
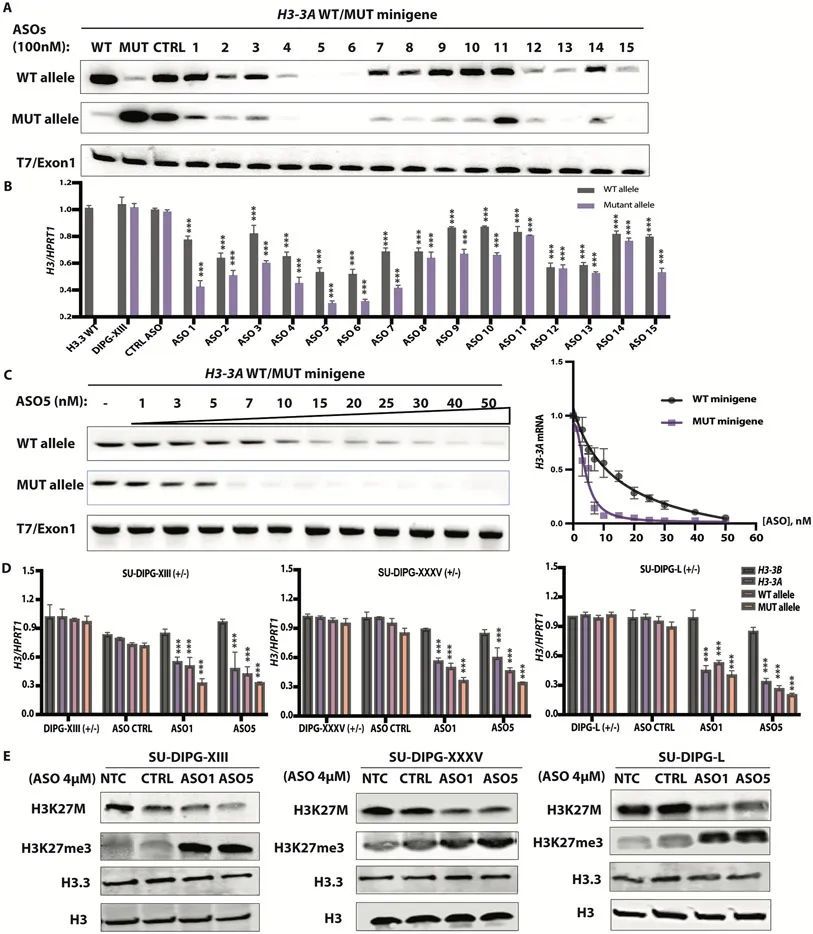
ASO介导的H3.3K27M消减延迟了神经球的生长并改变了细胞形态。
使用多个H3.3K27M DIPG患者衍生的细胞系作为神经球培养,作者观察到与乱序对照ASO相比,主导的ASO(ASO1和5)明显延缓了肿瘤细胞的生长。作者观察到,使用4μM ASO1或ASO5处理的SU-DIPG-XIII、SU-DIPG-XXXV和SU-DIPG-L神经球的增殖明显减慢(P < 0.001)(图2A、B和C)。相比之下,对于对照的H3.3WT胶质瘤细胞系,乱序对照ASO和ASO5对增殖没有可测量的影响。在第5天时点,这三个DIPG患者衍生的细胞系的细胞形态发生了变化,观察到了类似神经突起的过程(图2D)。这种形态学变化与作者在CRISPR敲除正位移植模型中观察到的分化表型一致。此外,使用主导的ASOs处理的SU-DIPG-XIII和SU-DIPG-L细胞系形成了较小的神经球(图2E)。
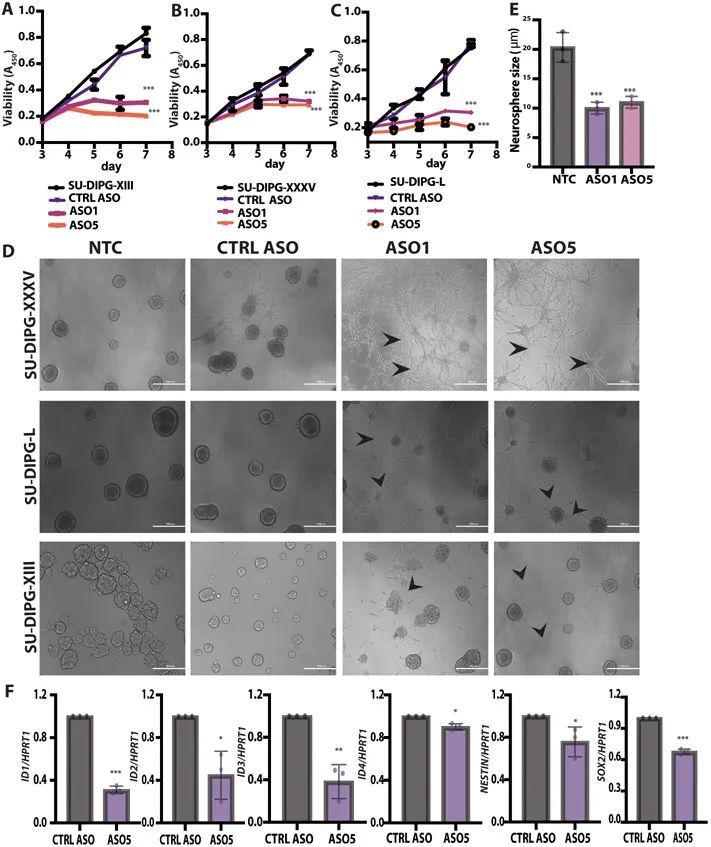
ICV注射铅ASO促进了H3.3K27M的消耗,在RCAS-Tva小鼠模型中降低了肿瘤级别和促进了分化
为了在体内测试作者的引导ASOs,作者首先采用了RCAS-Tva系统建立了小鼠DIPG模型。RCAS,即具有剪接受体的复制能力的鸟肉瘤-白血病病毒长末端重复序列(LTR),是一种只感染表达鸟Tva逆转录病毒受体的细胞的病毒载体(13)。此系统先前被用于显示小鼠组蛋白H3.3K27M或H3.1K27M加速胶质瘤发生,与另一个遗传小鼠模型的结果一致(14-16)。作者通过引入人H3-3A cDNA来改进该系统,其转录物可以被作者的人类特异性ASOs靶向。作者将产生编码P1噬菌体Cre重组酶、人H3.3K27M、小鼠血小板源性生长因子B(PDGFB)和萤火虫荧光素的鸡DF1细胞输送到由小鼠Nestin启动子驱动的带有Tva和转化相关蛋白53(Trp53)-floxed等位基因的新生小鼠的脑干中(图3A)。小鼠在3-6周内发展出高级别的胶质瘤,局部位于中线区域。通过组织学分析,作者发现FLAG标记的H3.3K27M存在于胶质瘤病变中。大约90%的H3.3K27M肿瘤显示全局H3K27me3减少,与正常相邻组织相比,呈现出与患者肿瘤组织学相似的模式。肿瘤细胞也呈阳性染色表达寡突胶质细胞谱系标记物寡突胶质细胞转录因子2(OLIG2)(15)。
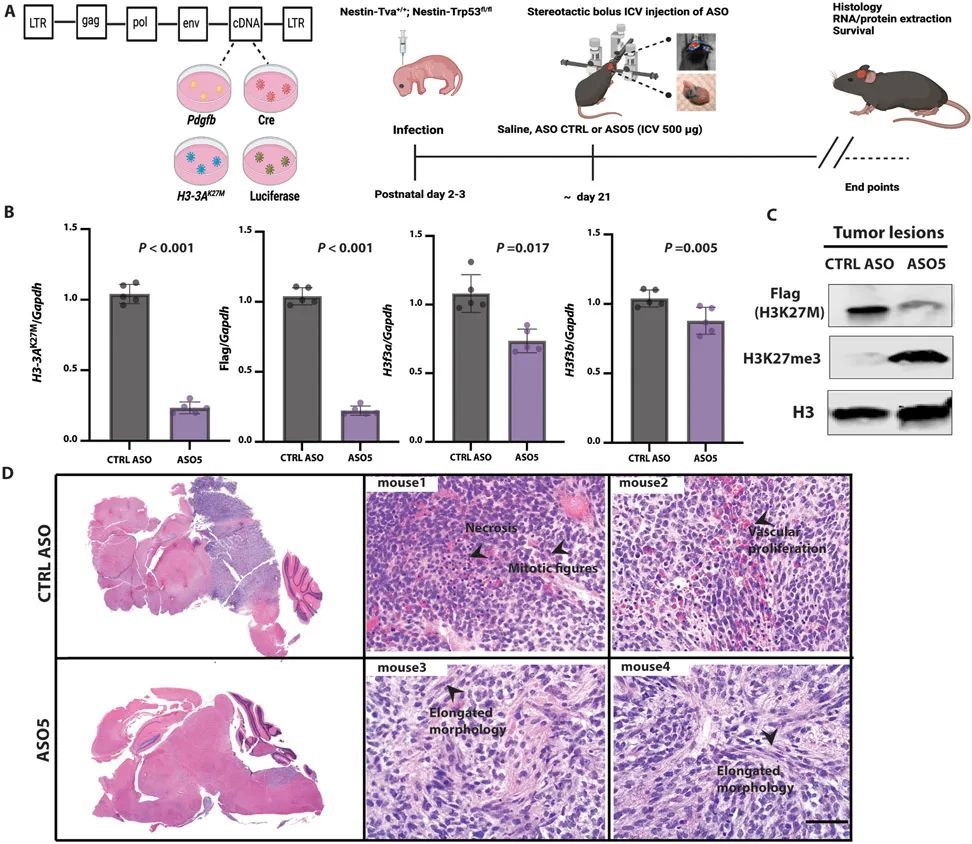
为了测试体内引物ASO的有效性,作者进行了立体定向ICV注射,将其直接输送到脑脊液中(CSF)(17, 18)。作者在肿瘤发生时,通过生物发光成像(图3A)检测到的时间点,向侧脑室注射了一次性剂量(500 μg)的引物ASO5或CTRL ASO溶于盐水中。作者在预设的时间点从肿瘤或正常相邻组织中提取RNA和蛋白质。ASO5显著(P < 0.001)降低了人类H3-3A K27M mRNA和FLAG标签的表达,对内源性小鼠野生型H3f3a的影响较小,但对H3f3b没有影响(图3B)。在蛋白质水平上,ASO5显著(P < 0.001)降低了人类FLAG标记的H3.3K27M,并增加了H3K27me3的丰度(图3C)。作者进一步观察到FLAG标记的免疫荧光染色的消失,即在约80%的处理细胞中,FLAG信号低于检测水平。
接受对照ASO处理的小鼠发展出高度增殖和侵袭性的胶质瘤,伴有大量有丝分裂图像、广泛的血管增生、偶尔的坏死以及坏死区周围的伪假性帕拉赛德。这些肿瘤没有包膜,边界不清晰,在肿瘤-脑界面处有一些肿瘤侵袭。相比之下,接受ASO5处理的小鼠显示出肿瘤生长的潜伏期延长,肿瘤呈现出延长的形态(图3D)。在这些肿瘤病变中,有丝分裂图像很少,坏死并不明显。此外,ASO5处理的肿瘤病变中的细胞形态学上类似于胶质细胞和成熟神经元。作者得出结论,体内由引导ASO引起的突变H3-3A的沉默导致了低级别肿瘤形成和更多分化的外观。
为了进一步描述组织学观察到的表型,作者对已知分化标记物进行了免疫荧光染色。在具有相同遗传背景但携带H3.3WT肿瘤的小鼠中,作者检测到成熟星形胶质细胞(GFAP)(19)、神经元(NeuN)(20)和少突胶质细胞(髓鞘碱性蛋白(MBP))(21)的标记物,表明H3.3WT胶质瘤中发生了神经发生和胶质发生(图4A)。相比之下,在存在H3.3K27M的肿瘤病灶中,检测到的GFAP、NeuN和MBP细胞较少(图4B,顶部面板)。经过一次脑室内注射ASO5剂量后,作者检测到大量的GFAP、NeuN和MBP细胞,与Ki67标记的增殖细胞数量减少相一致,Ki67是一个在细胞周期的所有活跃阶段表达的核细胞增殖相关抗原(图4B,底部面板)。Ki67的减少和GFAP、NeuN标记的增加在统计学上具有显著性(P = 0.002或0.003)(图4C和D)。作者通过对从正常相邻组织和肿瘤病灶中提取的总蛋白进行免疫印迹验证了这些结果(图4E)。作者得出结论,H3.33K27M阻碍了星形胶质细胞和神经元的分化,而ASO介导的H3.3K27M消耗恢复了分化程序。

ICV注射ASO5降低了NESTIN细胞群体,并延长了肿瘤生长的潜伏期
DMG在特定的时空背景下出现,并在中童期发生,发病高峰年龄为6至9岁。细胞起源和微环境对肿瘤生长至关重要(26, 27)。先前的回顾性克隆分析显示,NESTIN + 细胞在人类中脑、脑桥和延髓中富集,贯穿整个童年期,且在腹侧脑桥处密度最高;因此,NESTIN + 细胞群体与DIPG的时空发病率完全一致(27)。在作者的小鼠模型中,相对于正常相邻脑组织,NESTIN + 细胞在肿瘤病变部位富集。经过ASO5治疗后,NESTIN + 细胞群体明显减少,与GFAP + 细胞数量的增加呈负相关(图4F)。此外,ASO5治疗的小鼠的存活时间显著延长(P < 0.0001),与对照组ASO治疗的小鼠相比(图4G)。这些结果表明,H3.3K27M肿瘤起源于神经干细胞(NESTIN + ),并与TRP53缺失和PDGF信号传导相关。
ASO5治疗在RCAS-Tva模型中产生的分化细胞是肿瘤起源的。
为了确定ASO5处理后出现的分化细胞是否来自肿瘤,而不是小鼠细胞被招募到损伤部位,作者利用在RCAS-Tva模型中表达的血凝素(HA)标记的Pdgfb和Cre cDNAs。作者使用HA抗体进行免疫荧光染色,并观察到大部分肿瘤细胞都是HA + 。当作者用HA和各个分化标记物双标记细胞时,作者观察到大量的HA + 星形胶质细胞、神经元和少突胶质细胞(图5A)。
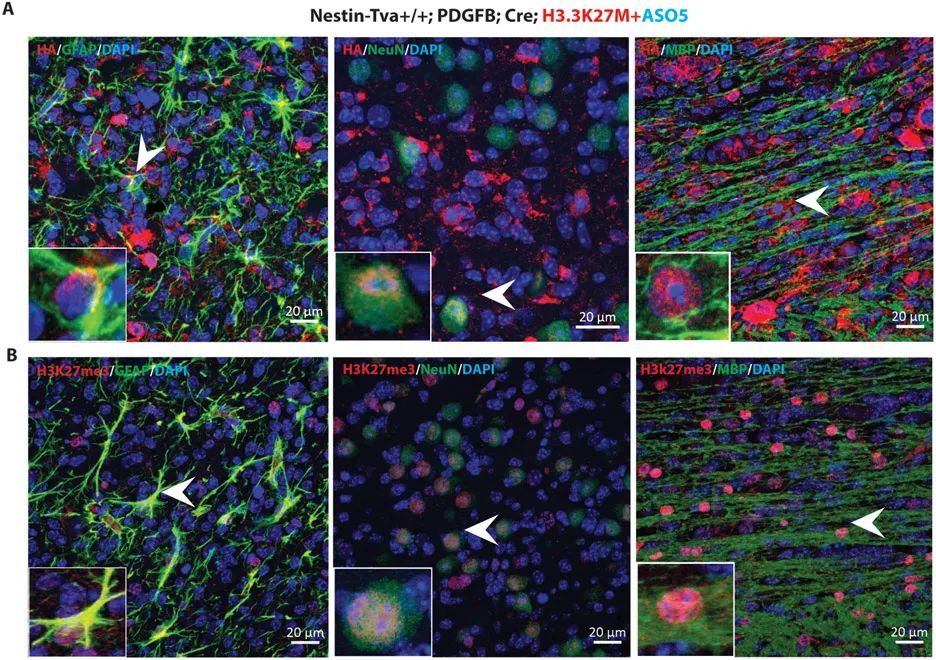
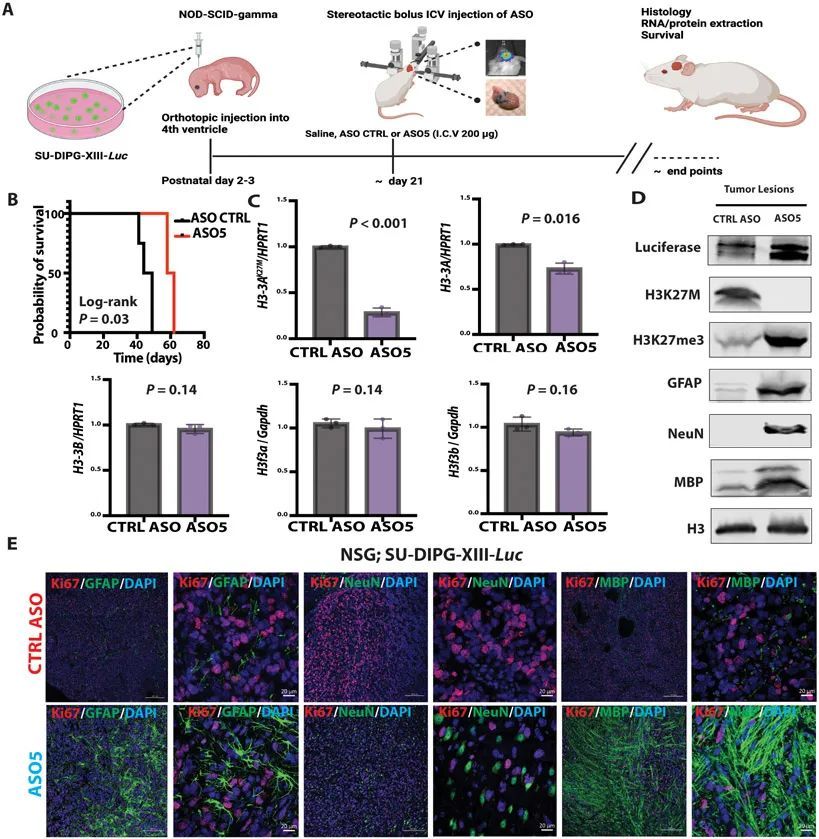
ICV注射ASO5促进了星形胶质细胞、神经元和少突胶质细胞的分化,并减少了DIPG患者源自移植模型的肿瘤增殖
为了确认和扩展上述观察结果,作者还在一个原位异种移植小鼠模型中测试了ASO5,如(9)所述,使用图3中显示的SU-DIPG患者系列之一。作者将10个表达荧光素的SU-DIPG-XIII细胞注射到P3免疫缺陷的NSG小鼠的第4脑室中,并使用生物发光成像来跟踪肿瘤发生。由于这些免疫缺陷小鼠无法耐受RCAS-Tva模型中使用的高浓度ASO,作者对其进行了单次脑室注射,注射200μg对照ASO或ASO5(图6A)。与对照ASO处理组相比,ASO5处理组的小鼠存活时间显著延长(P = 0.03)(图6B)。与RCAS-Tva小鼠模型类似,ASO5显著(P < 0.001;P = 0.016)降低了人类H3-3A mRNA和总人类H3-3A mRNA的表达,但不影响内源性小鼠野生型H3f3a或H3f3b(图6C)。此外,ASO5降低了人类H3.3K27M蛋白的表达,并增加了H3K27me3修饰的程度。ASO5处理还导致Luciferase标记的SU-DIPG-XIII细胞系中GFAP、NeuN和MBP蛋白的丰度升高(图6D)。ASO5处理的小鼠在肿瘤病灶中表现出不同的表型(GFAP + ,NeuN + ,和MBP + ),细胞增殖较少(Ki67 + ),而在对照组ASO处理的小鼠中,神经发生和胶质发生受到损害,大部分肿瘤细胞仍处于增殖状态(图6E)。与RCAS-Tva模型类似,表达各种分化标记的细胞也具有H3K27me3 + 。
总结
总之,作者开发了在体外和体内直接靶向癌组蛋白mRNA的gappmer ASOs。前导ASO有效降解H3-3AK27M mRNA,减少患者来源细胞和小鼠模型中的H3.3K27M蛋白。H3.3K27M蛋白丰度的降低导致小鼠模型中神经发生和胶质发生的恢复,肿瘤生长的潜伏期延长,存活率增加。药物干预的效果不如预期的完全基因敲除,因为ASO治疗减少但不会消除突变蛋白的表达。此外,与植入前基因敲除不同,治疗性ASO是在肿瘤发作后给药的,这代表了一种更现实的临床情况。作者预计,最大的临床疗效可能需要联合治疗,包括ASO给药,例如放疗(36)或CAR-T细胞免疫疗法(37,38)。
这篇关于H3.3K27M弥漫性中线胶质瘤的反义寡核苷酸治疗的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!