本文主要是介绍Single-cell 10x Cell Ranger analysis,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
first step download SRR data
#这是批量下载
nohup prefetch -X 100GB --option-file SRR_Acc_List.txt &
nohup fastq-dump --gzip --split-files -A ./SRR13633760 -O /home/scRNA/ &
next Build a custom reference using Cell Ranger mkref
首先,找到您物种的参考基因组 FASTA 和 GTF 文件。如果该物种可从 Ensembl 数据库中获得,我们建议使用那里的文件。来自 Ensembl 的 GTF 文件包含可选标签,使过滤变得容易。如果 Ensembl 无法获得您感兴趣的物种,也可以使用其他来源的 GTF 和 FASTA 文件。请注意,GTF 文件是必需的,而不支持 GFF 文件。(请参阅 GFF/GTF 文件格式 - 定义和支持的选项)
这个是 Ensembl 的链接,进行选取物种进行制作参考基因组文件
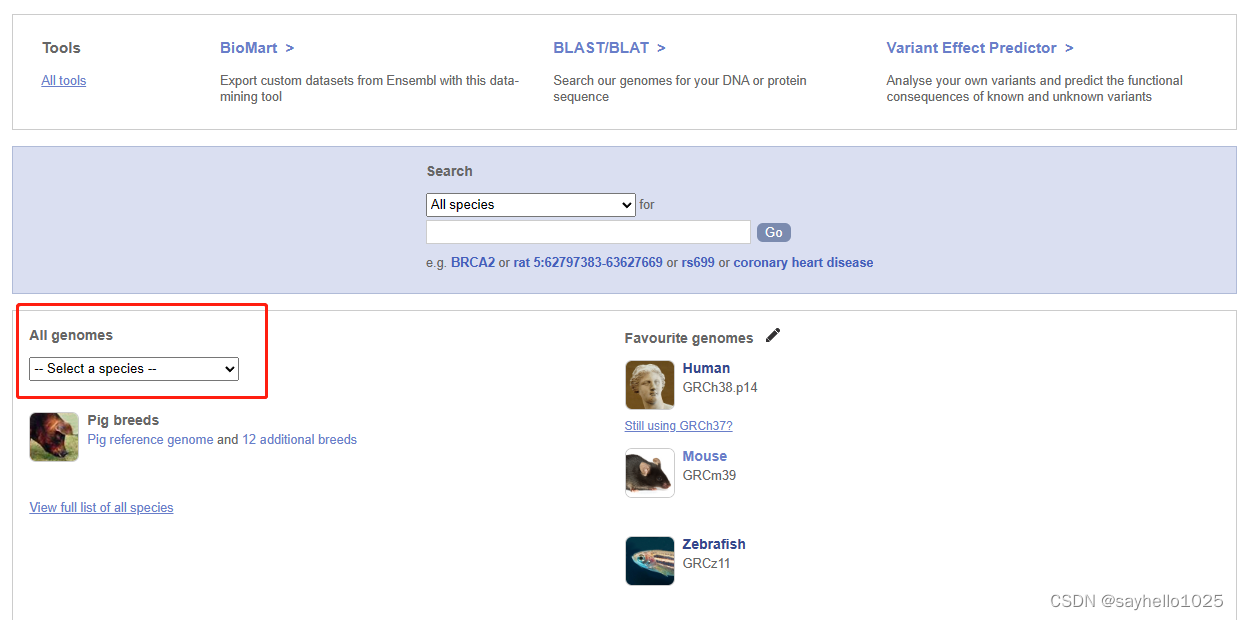
这里以大鼠进行演示
分别打开图中的红色框中的内容
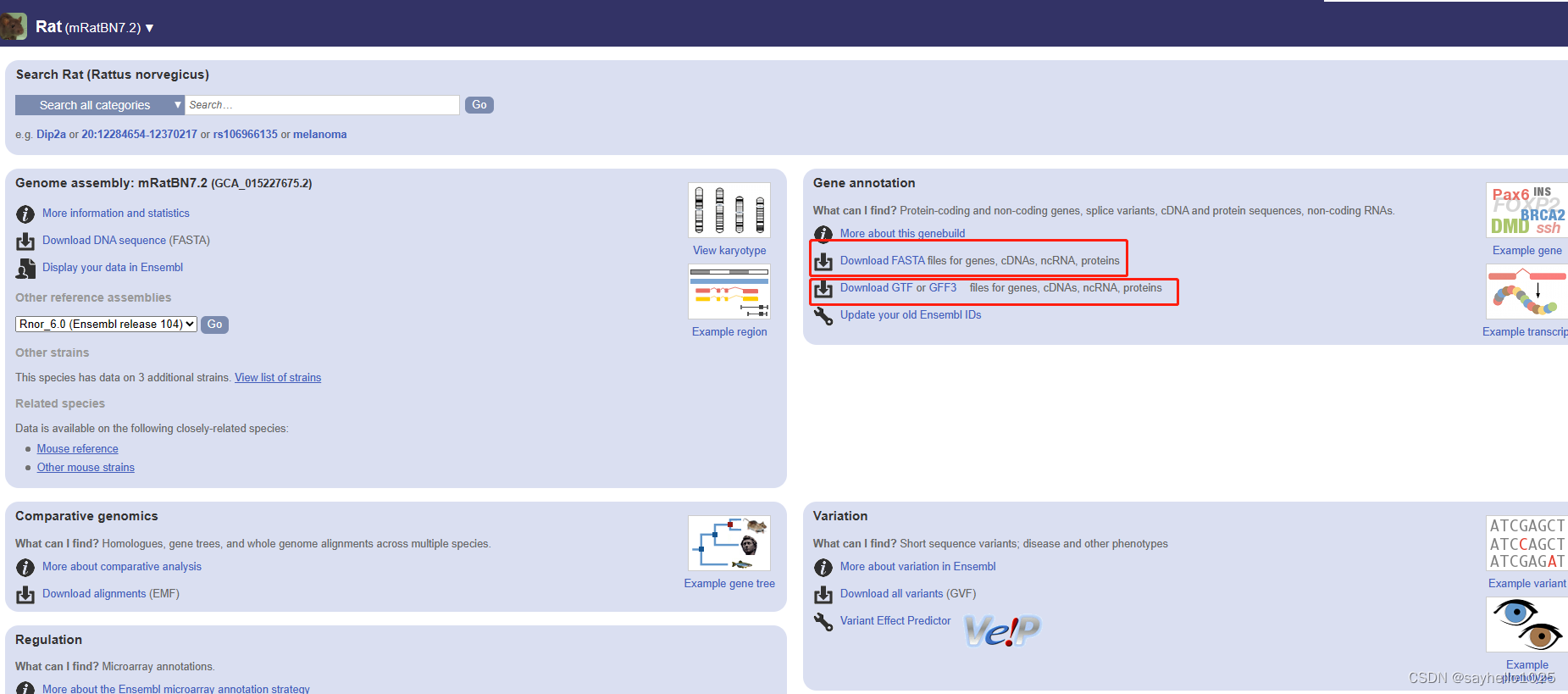
点击红色框中的内容
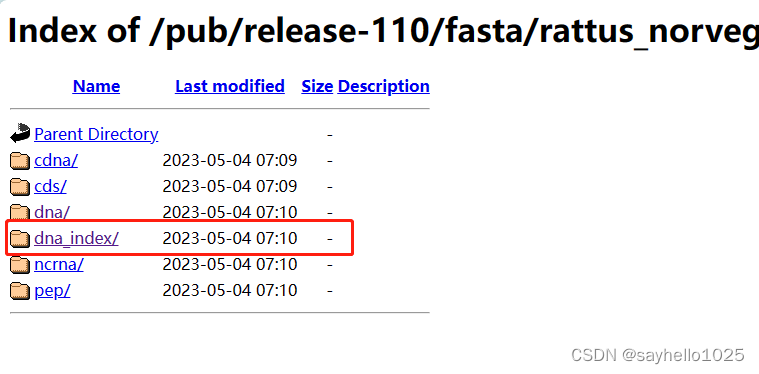
选取选择了顶级的FASTA文件下载
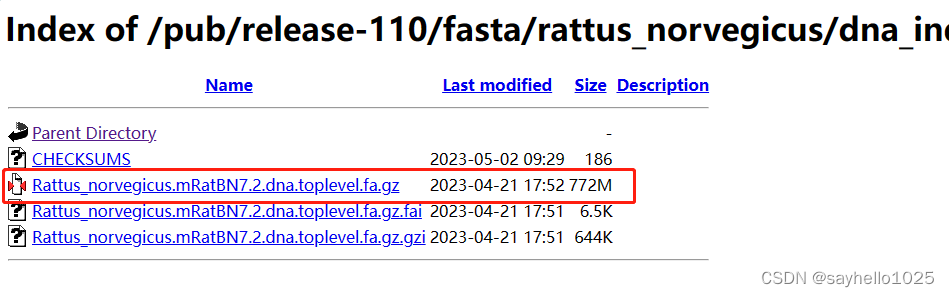
到这里已经完成第一个文件的下载,下面展示第二个文件下载,需要打开下面图的框,选择Download GTF
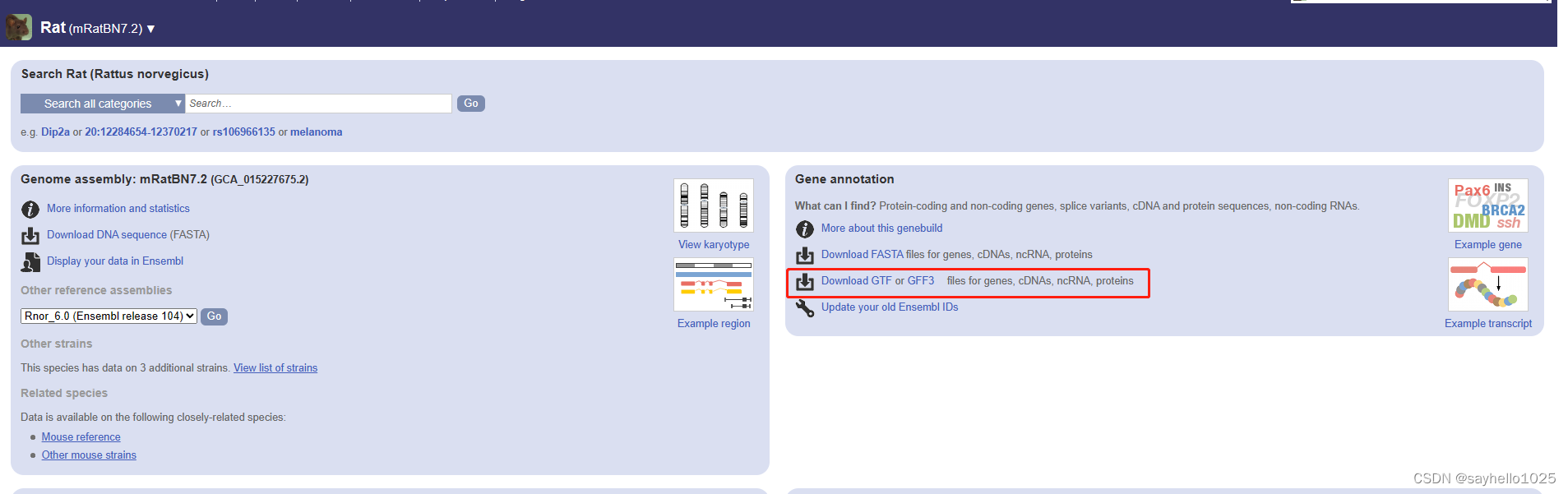
选择这个下载
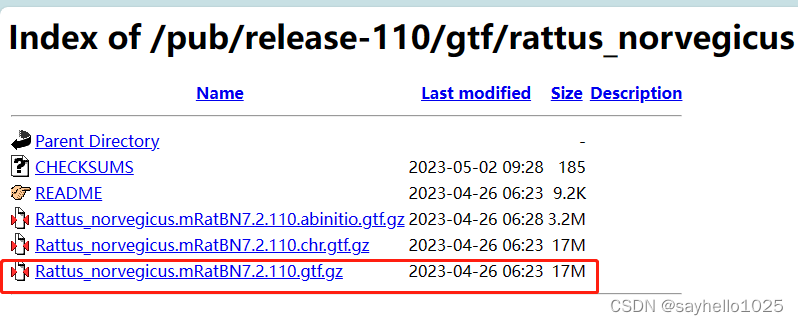
至此我们全部完成下载内容
接下来进行构建参考基因组
这一步不是在我理解不是非必要,在做一些分析时,可以根据这个排除以基因,例如 仅添加–attribute=gene_biotype:protein_coding 只做编码蛋白在所有细胞中的分析
看一下说明
(base) hwsw@shpc-2596-instance-GkVAxmvG:~$ $cellranger mkgtf
Usage:mkgtf <input_gtf> <output_gtf> [--attribute=KEY:VALUE...]mkgtf -h | --help | --version
(base) hwsw@shpc-2596-instance-GkVAxmvG:~$ $cellranger mkgtf -h
Genes GTF tool for 10x Genomics Cell Ranger.Filter user-supplied GTF files for use as Cell Ranger-compatible
genes files for mkref tool.The commands below should be preceded by 'cellranger':Usage:mkgtf <input_gtf> <output_gtf> [--attribute=KEY:VALUE...]mkgtf -h | --help | --versionArguments:input_gtf Path to input genes GTF file.output_gtf Path to filtered output genes GTF file.Options:--attribute=<key:value>Key-value pair in attributes field to be kept in the GTFfile.-h --help Show this message.--version Show version.
这个是选择想要的表型然后进行过滤
#Filter GTF
cellranger=/home/hwsw/cellranger-7.1.0/cellranger
$cellranger mkgtf \
Rattus_norvegicus.mRatBN7.2.105.gtf Rattus_norvegicus.mRatBN7.2.105.filtered.gtf \
--attribute=gene_biotype:protein_coding \
--attribute=gene_biotype:lncRNA \
--attribute=gene_biotype:antisense \
--attribute=gene_biotype:IG_LV_gene \
--attribute=gene_biotype:IG_V_gene \
--attribute=gene_biotype:IG_V_pseudogene \
--attribute=gene_biotype:IG_D_gene \
--attribute=gene_biotype:IG_J_gene \
--attribute=gene_biotype:IG_J_pseudogene \
--attribute=gene_biotype:IG_C_gene \
--attribute=gene_biotype:IG_C_pseudogene \
--attribute=gene_biotype:TR_V_gene \
--attribute=gene_biotype:TR_V_pseudogene \
--attribute=gene_biotype:TR_D_gene \
--attribute=gene_biotype:TR_J_gene \
--attribute=gene_biotype:TR_J_pseudogene \
--attribute=gene_biotype:TR_C_gene
准备单细胞分析参考基因组
cellranger mkref --help 查看命令参数
Reference preparation tool for 10x Genomics Cell Ranger.Build a Cell Ranger-compatible reference folder from user-supplied genome FASTA and gene GTF files. Creates a new folder named after the genome.The commands below should be preceded by 'cellranger':Usage:mkref--genome=NAME ...--fasta=PATH ...--genes=PATH ...[options]mkref -h | --help | --versionArguments:genome #输出文件夹 Unique genome name(s), used to name output folder[a-zA-Z0-9_-]+. Specify multiple genomes byspecifying the --genome argument multiple times; theoutput folder will be <name1>_and_<name2>.fasta #FASTA参考基因组绝对路径 Path(s) to FASTA file containing your genome reference.Specify multiple genomes by specifying the --fastaargument multiple times.genes #.filtered.gtf注释文件绝对路径Path(s) to genes GTF file(S) containing annotated genesfor your genome reference. Specify multiple genomesby specifying the --genes argument multiple times.Options:--nthreads=<num> This option is currently ignored due to a bug, and will be re-enabledin the next Cell Ranger release.--memgb=<num> Maximum memory (GB) used when aligning reads with STAR.Defaults to 16.--ref-version=<str> Optional reference version string to include withreference.-h --help Show this message.--version Show version.
进行分析时间很长花费2-3小时
#Run mkref
cellranger mkref \
--genome=mRatBN7 \
--fasta=Rattus_norvegicus.mRatBN7.2.dna.toplevel.fa \
--genes=Rattus_norvegicus.mRatBN7.2.110.filtered.gtf \
--ref-version=1.0.0
我截取别人图运行截图
Apr 15 14:36:45 ..... started STAR run
Apr 15 14:36:45 ... starting to generate Genome files
Apr 15 14:38:52 ... starting to sort Suffix Array. This may take a long time...
Apr 15 14:39:03 ... sorting Suffix Array chunks and saving them to disk...
Apr 15 16:40:45 ... loading chunks from disk, packing SA...
Apr 15 16:41:47 ... finished generating suffix array
Apr 15 16:41:47 ... generating Suffix Array index
Apr 15 16:46:07 ... completed Suffix Array index
Apr 15 16:46:07 ..... processing annotations GTF
Apr 15 16:46:19 ..... inserting junctions into the genome indices
Apr 15 16:55:08 ... writing Genome to disk ...
Apr 15 16:55:23 ... writing Suffix Array to disk ...
Apr 15 16:56:00 ... writing SAindex to disk
Apr 15 16:56:08 ..... finished successfully
Creating new reference folder at /home/hanjiangang/single_Cell/example/ref/ovis_aries/ovis_aries
...doneWriting genome FASTA file into reference folder...
...doneIndexing genome FASTA file...
...doneWriting genes GTF file into reference folder...
...doneGenerating STAR genome index (may take over 8 core hours for a 3Gb genome)...
...done.Writing genome metadata JSON file into reference folder...
Computing hash of genome FASTA file...
...doneComputing hash of genes GTF file...
...done...done>>> Reference successfully created! <<<
You can now specify this reference on the command line:
cellranger --transcriptome=/home/hanjiangang/single_Cell/example/ref/ovis_aries/ovis_aries ..
这里面最后会生成–transcriptome=/home/hmsw/Rattus 这个文件夹
直接进行10x的标准流程
/home/hwsw/cellranger-7.1.0/cellranger count --id=SRR19145616 \
--transcriptome=/home/hwsw/Rattus \
--fastqs=/home/scRNA/SRR19145616 \
--sample=SRR19145616 \
--localcores=30 \
--localmem=300 \
--nosecondary
这篇关于Single-cell 10x Cell Ranger analysis的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!


![[LeetCode] 137. Single Number II](/front/images/it_default.jpg)