本文主要是介绍孟德尔随机化--研究,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
文章目录
- 写在前面
- 为什么要做孟德尔随机
- 原理
- 第三需要满足的假设条件
- 方法
- IVW与Egger
- 方法使用原则
- F
- 结果解读
- 1.总体结果
- 如何下载数据
- 1.GWAS catelog
- 2.ieu数据库
- 3.ukb数据库
- 4.芬兰数据库
- R代码实现
- 基本的暴露结局分析
- 敏感性分析(分析结果完之后看结果的可靠性)
写在前面
看了一些孟德尔随机化内容,大概整理一下步骤
参考博文:
第一篇:https://mp.weixin.qq.com/s/YdGgCi-hSLpBP3ngA_SvpQ
第二篇:https://zhuanlan.zhihu.com/p/582635108
第三系列:有何AI与医学(公众号)–孟德尔随机化研究系列
视频:B站https://www.bilibili.com/video/BV1EM4y1f7F6/?spm_id_from=autoNext&vd_source=74afe31b29c85e8f5a1afbc0d0ec17f0
系列:https://mp.weixin.qq.com/s/zo1nv3nlcwI4D8Lu0bXzMQ
多效性的讲解:https://zhuanlan.zhihu.com/p/439016497
为什么要做孟德尔随机
1.我们需要因果性分析,而不是相关性分析
2.现实中前瞻性比较难做到,而我们已有很多的研究数据可以用来做因果性推断
3.与GWAS的联系?需要用到GWAS的数据,GWAS算是只分析基因突变位点与表型的关系,孟德尔随机追寻的是暴露因素与表型的联系
4.类似于随机对照实验,随机实验以随机分组作为入组,孟德尔随机则是以位点突变作为入组。
5.非遗传变异的变量相关性较强,解释了为什么要选遗传变量作为工具变量
6.为什么数据最好是单人种,因为由于人口分层而产生偏见。就是说所有既能影响到工具变量又能影响结果的因素都应该控制。
原理
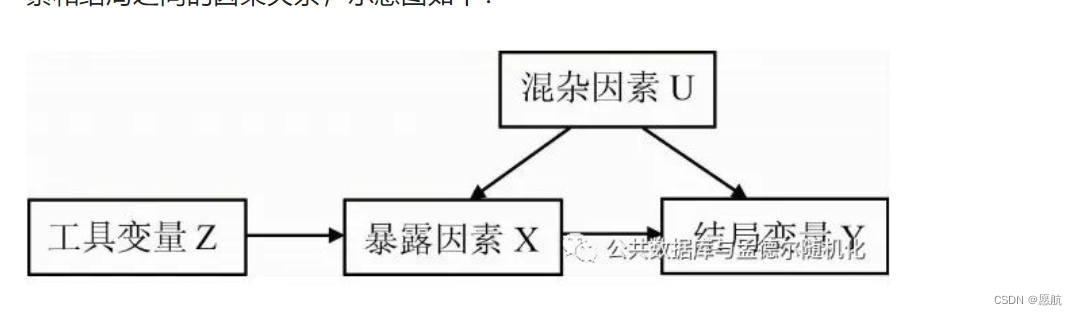
工具变量是基因型,暴露因素是表型,结局变量是响应变量。举个例子,研究饮酒量(暴露因素)引起CHD发病(结局变量)的风险,ALDH2基因多态性(工具变量)决定血中乙醛浓度(中间变量1),后者可影响饮酒行为(中间变量2),从而改变饮酒量(暴露因素),所以血乙醛浓度这一中间表型可间接代表饮酒量(该例取自第一篇博文)。
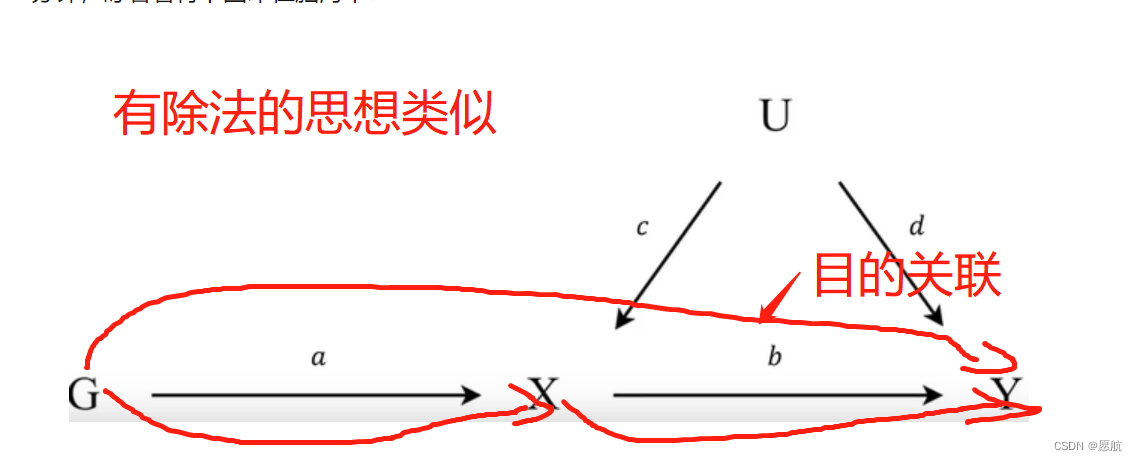
同时,第二篇博文提出,研究目的是得到XY之间关联,但通过Z-X-Y/Z-X,事实肯定不是简单的除法。这么做的原因,大致是因为有混杂因素存在,XY之间关联不好直接算。补上第二篇博文参考图:
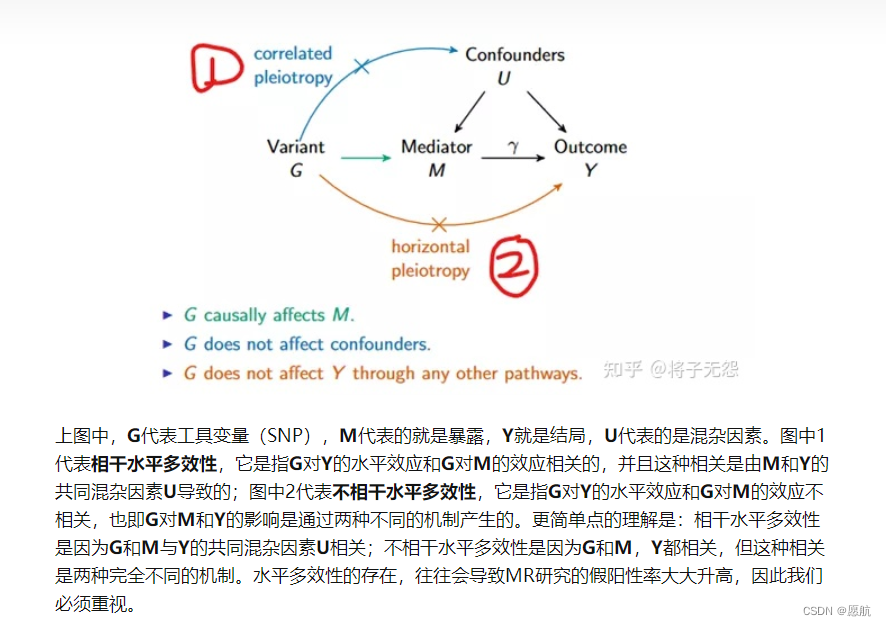
何为多效性(违背下面2、3原理)
水平基因多效性指2
垂直基因多效性指
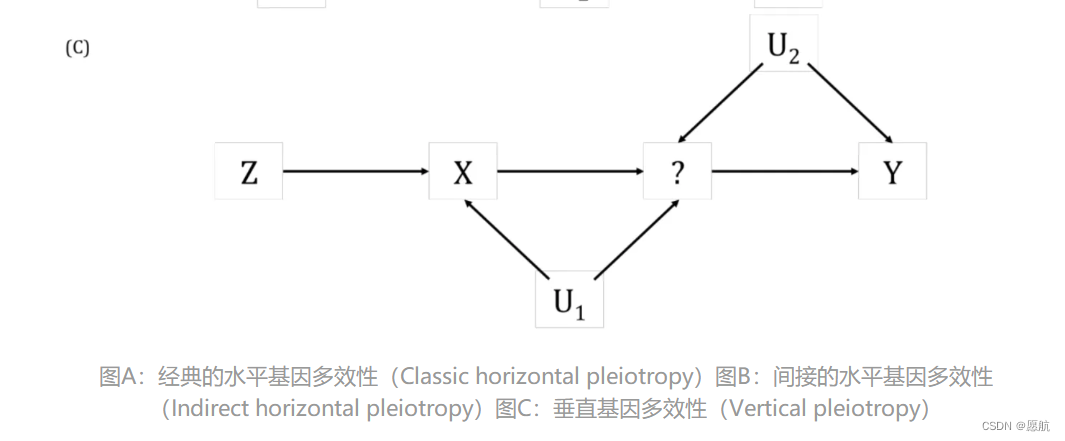
第三需要满足的假设条件
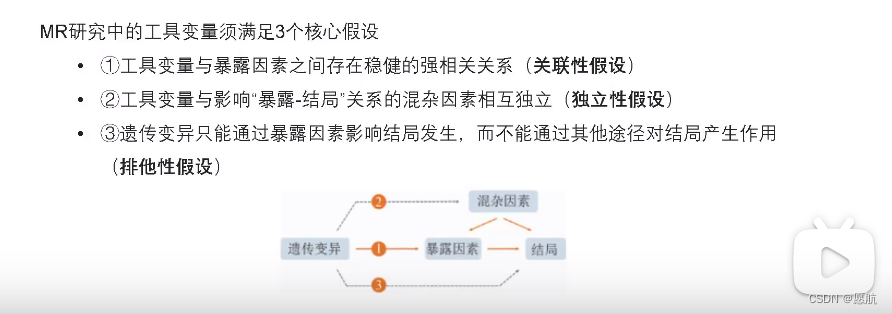
关于第二点独立性,一般是指不同暴露下的其它混杂因素分布大致相同
1.强工具变量F值大于10
2.暴露因素与结局变量的人群还不应该交叉,而且不推荐亚洲人,因为比如亚洲人中不同种族的差异很明显(比如东亚人和中亚人)
3.在孟德尔随机化中,使用某种特定遗传变异作为IV的任何理由都应以生物学知识而非统计学检验为基础。
方法
IVW与Egger
在IVW的假设中,我们认为这些SNP(也称IV)是没有多效性的,同时考虑到GWAS的结果多为表型标准化后做出来的,所以我们认为结局和暴露之间是正比例关系。因此,我们在使用IVW方法必须要保证这些SNP没有多效性,否则结果会有很大的偏倚。
在MR-Egger的假设中,我们考虑截距项的存在,并用它来评估多效性。如果该截距项和0非常接近,那么MR-Egger回归模型就和IVW非常接近,但是如果截距项和0相差很大,那就说明这些IV间可能有水平多效性存在。
IVW是最早使用的方法,也是最常用的,它需要SNP完全符合MR研究三原则才能得到正确的因果估计;MR-Egger多加了截距项,其主要目的是判断水平多效性的有无;Weighted Median是利用大部分SNP(majority of genetic variants)来判断因果关系的有无。
方法使用原则

F

结果解读
1.总体结果

暴露与结果是否有因果因素,所以不会出现具体SNP,snp的数量在nsnp上,mydata上有时会删掉部分基因列mydata$mr_keep
如何下载数据
1.GWAS catelog
https://www.ebi.ac.uk/gwas/downloads/summary-statistics,这个网址,但可能需要翻梯子
2.ieu数据库
https://gwas.mrcieu.ac.uk/,这个貌似不需要开梯子
3.ukb数据库
https://www.nealelab.is/uk-biobank,貌似需要开梯子
4.芬兰数据库
https://www.finngen.fi/en/access_results
上述数据库具体操作可详见https://www.bilibili.com/video/BV1wV4y1w7w6/?spm_id_from=333.788&vd_source=74afe31b29c85e8f5a1afbc0d0ec17f0教学视频
R的twosampleMR能下载部分数据,其中以available_outcomes()查看所有下载路径,再查看包含的所有数据库

R代码实现
基本的暴露结局分析
# 基本的暴露结局分析
library(TwoSampleMR)
# 一般下面提取暴露变量,考虑到连锁不平衡时需要r2=0.001,kb=10000。去掉在10000kb范围内与最显著SNP的r2大于0.001的SNP;
bmi_exp <- extract_instruments(outcomes='ieu-a-835',clump=TRUE, r2=0.01,kb=5000,access_token= NULL
)
dim(bmi_exp)
# [1] 80 15
t2d_out <- extract_outcome_data(snps=bmi_exp$SNP,outcomes='ieu-a-26',proxies = FALSE,maf_threshold = 0.01,access_token = NULL
)
dim(t2d_out)
# [1] 80 16mydata <- harmonise_data(exposure_dat=bmi_exp,outcome_dat=t2d_out,action= 2
)res <- mr(mydata)
res
敏感性分析(分析结果完之后看结果的可靠性)

异质性检验(Q_pval远小于0.05具有异质性),可剔除某些outcome的P值非常小的SNP,或可使用随机效应模型观察结局校正后是否还有显著性
het <- mr_heterogeneity(mydata)
het
mr(mydata,method_list=c('mr_ivw_mre')) #使用随机效应模型
多效性检验(p>0.05说明没有多效性,不需要处理)
pleio <- mr_pleiotropy_test(mydata)
pleio
逐个剔除检验
single <- mr_leaveoneout(mydata)
mr_leaveoneout_plot(single)
不同方法绘图
mr_scatter_plot(res,mydata)
#
森林图
res_single <- mr_singlesnp(mydata)
mr_forest_plot(res_single)
漏斗图
mr_funnel_plot(res_single)
备用代码
#设置工作路径
#第一次使用,下载TwoSampleMR包,将下方俩行#去掉后分别运行
#install.packages("remotes")
#remotes::install_github("MRCIEU/TwoSampleMR")
#读取TwoSampleMR包
library(TwoSampleMR)#######找中间变量######
bmi_exp <- extract_instruments(outcomes = 'ieu-b-4877')
dim(bmi_exp) #查看筛选出多少个SNP
head(bmi_exp) #查看前五行信息#######利用汇总数据库ID进行结局SNP提取#######
t2d_out <- extract_outcome_data(snps = bmi_exp$SNP,#根据暴露SNP编号在结局中查找对用SNPoutcomes = "ebi-a-GCST006906",#查找数据库ID号为ebi-a-GCST006906的SNPproxies = TRUE,#在结局中找不到的SNP是否用1000组的代替,默认TRUE,R方0.8maf_threshold = 0.01,#最小等位基因频率access_token = NULL#是否通过谷歌访问,国内选NULL
)#查看暴露SNP基本情况,发现少了几个,
#一般可能不符合条件,或者再结局中没有找到
dim(t2d_out) #查看筛选出多少个SNP
head(t2d_out) #查看前五行信息
######合并暴露数据与结局数据#######
mydata <- harmonise_data(exposure_dat=bmi_exp,outcome_dat=t2d_out,action= 2#是否去除回文序列,2去除,1不去除,一般我们要去除
)
#如果需要保存SNP信息,则去掉write前#运行
#write.table(mydata, file="mydata.xls",sep="\t",quote=F)
#数据已经准备完成,接下来开始做MR分析
## MR
res <- mr(mydata) #一般看IVW行,如果p《0.05,说明有潜在的因果关系。
#是首选项,其他结果是辅助
## Heterogeneity statistics
mr_heterogeneity(mydata) # Q_pval<0.05说明有异质性,我们需要同质性样本
#当为异质性样本时,需要根据漏斗图考虑删掉部分SNP,再重新拟合
# 异质性可视化,所谓的漏斗图
mr_forest_plot(singlesnp_results = mr_singlesnp(mydata))
# pleiotropy test
mr_pleiotropy_test(mydata)# leave-one-out analysis,一个个剔除,每个变量应该都在0右边,且偏差不大
res_loo <- mr_leaveoneout(mydata)mr(mydata, method_list=c("mr_egger_regression", "mr_ivw"))########MR分析##########
res_mi=generate_odds_ratios(mr(mydata))
#我们这里为方便直接完成转化OR所需结果就都涵盖在内
res_mi#输出查看结果
#保存MR结果
write.table(res_mi, file="res_mi.xls",sep="\t",quote=F)# scatter plot
dat <- mydata
p1 <-mr_scatter_plot(res, dat)
length(p1) # to see how many plots are there
p1[[1]]# forest plot
res_single <- mr_singlesnp(dat)#,all_method=c("mr_ivw", "mr_two_sample_ml")) to specify method used
p2 <- mr_forest_plot(res_single)
p2[[1]]## funnel plot for all
p3 <- mr_funnel_plot(res_single)
p3[[1]]
mr_report(dat)
out <- directionality_test(dat)
kable(out)
mr_steiger(p_exp = dat$pval.exposure, p_out = dat$pval.outcome, n_exp = dat$samplesize.exposure, n_out = dat$samplesize.outcome, r_xxo = 1, r_yyo = 1,r_exp=0)从ebi下载数据,并根据文件分割出所需要的GWAS数据
setwd("E:\\xianyu\\MR")
data <- vcfR::read.vcfR("bbj-a-140.vcf.gz") #读取VCF文件
gt <- data.frame(data@gt)#ES代表beta值、SE代表se、LP代表-log10(P值)、AF代表eaf、“ID”代表SNP的ID
library(limma)
dat <- strsplit2(gt$bbj.a.140, split = ":")#strsplit切分;unlist解开
fix<-data.frame(data@fix)#为SNP位点的基本信息frame<-data.frame(dat)
colnames(frame)<-c("ES","SE","LP","AF","ID")
frame$rs = fix$ID
write.table(frame, file=" pancreatic_cancer.txt",sep="\t",quote=F)
ggadjustedcurves这篇关于孟德尔随机化--研究的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!







