本文主要是介绍CNV学习3(CNV软件使用学习--input),希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
PennCNV 输入文件都是文本格式。它需要信号强度文件、 HMM 文件、 PFB 文件和可选的 GCModel 文件。用户将需要准备正确的格式的信号强度文件,而所有其他文件已经捆绑在 PennCNV 软件包。下面我们描述了各种输入文件格式,并描述了从各种来源准备信号强度文件的过程。(抓重点:信号强度文件最需要自己准备!!!)
输入信号强度文件是一个文本文件,其中包含每行一个SNP的信息,并且每行中的所有字段都用制表符分隔。以下是一个例子:
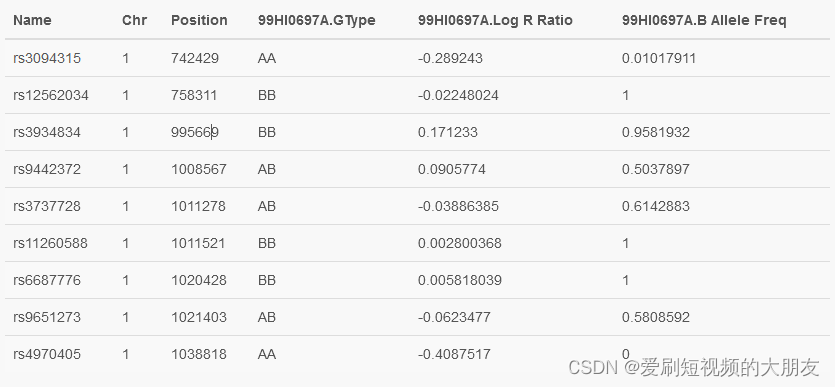 文件的第一行指定了每个制表符分隔的列的含义。例如,文件中每行有6个字段,分别对应 SNP 名称、染色体、位置、基因型、对数 R 比率(LRR)和等位基因频率(BAF)。
文件的第一行指定了每个制表符分隔的列的含义。例如,文件中每行有6个字段,分别对应 SNP 名称、染色体、位置、基因型、对数 R 比率(LRR)和等位基因频率(BAF)。
CNV 调用只需要 SNP Name、 LRR 和 BAF 值(染色体坐标在 PFB 文件中注释,见下文)。因此,下面的输入信号强度文件也可以工作。请注意,LRR 和 BAF 的相对位置与前一个文件不同; 同样,头行告诉程序第二列代表 BAF 值,而第三列是 LRR 值。
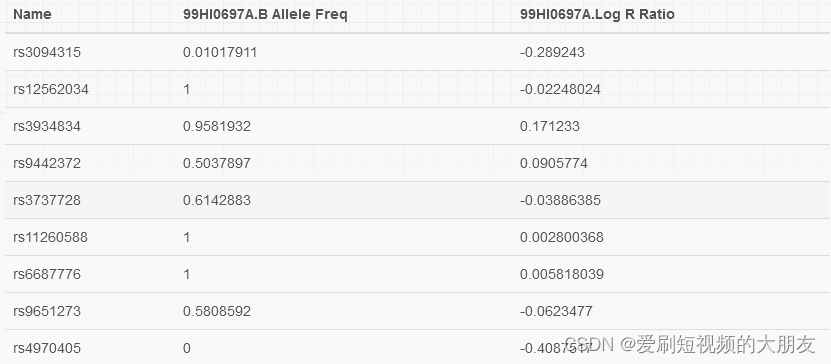
从 Illumina 报告中准备输入信号强度文件
有时,用户可能会从基因分型中心得到一个文本文件,其中应该包含一个给定项目中所有样本中所有标记的信号强度值(对数 R 比率和 B 等位基因频率)。前几行应如下:
ID、 B Allele Freq 和 Log R Ratio,那么有必要联系数据提供者,并要求他们重新生成一个包含所有这些必需字段的报告文件。在文件中包含额外的字段,比如 X、 Y、 X Raw 和 Y Raw 字段,这很好,但是只有 LRR 和 BAF 值对 CNV 调用有用。

提示: 报告文件可以直接从 BeadStudio 项目文件中生成,点击分析菜单,选择 Report,然后选择 Final Report,然后确保从可用字段拖动 Log R Ratio 和 B Allele Freq 字段到显示字段,这样这两个信号强度测量就可以导出到最终报告文件中。或者,从显示字段中删除所有其他垃圾字段,如 GType、 GC 得分、 X Raw 等,以加快处理速度并减小文件大小。
PennCNV 软件包提供了一个方便的脚本,可以将这个长而庞大的报告文件转换为单独的信号强度文件,以便与 PennCNV 呼叫一起使用,每个文件包含一个样本的信息。
[kaiwang@cc ~/]$ split_illumina_report.pl -prefix rawdata1/ M00290_ Ballel_logR.txt
NOTICE: Writting to output signal file rawdata1/KS2231000715
NOTICE: Writting to output signal file rawdata1/KS2231007035NOTICE: Writting to output signal file rawdata1/KS3205046076
NOITCE: Finished processing 2193946 lines in report file M00290 _Ballel_logR.txt, and generated 48 output signal intensity files
输出文件名对应于原始报表文件中的 SampleID 字段。在上面的命令中,-prefix 参数用于指定文件名的前缀(在本例中,将文件保存到 rawdata1/ directory)。我们可以用-suffix .txt 在文件名中添加后缀。此外,我们可以使用-numeric 参数,以便输出文件名为 split1、 split2、 split3格式。在某些情况下,基因分型中心可能以逗号分隔的格式而不是制表符分隔的格式提供报告文件; 在这种情况下,可以使用-comma参数来处理这种文件。
从 BeadStudio 项目文件中准备输入信号强度文件
在某些情况下,我们可以获得 BeadStudio 项目文件格式的基因分型数据(有时也可以获得很多 idat 格式的文件,这些文件可以构建到单个项目文件中)。本节的目标是说明如何从基因分型项目导出信号强度数据(预计算的 Log R Ratio 和 B Allele Freq 数据)到文本文件,以便随后可以由 PennCNV 进行分析。我们需要一个带有 Illumina BeadStudio 软件的计算机系统,可以从你的微阵列核心设施中获得。
对于本教程,我们可以将教程项目文件下载为一个约100MB 大小的、单独的 tutorial_beadstudio.zip 文件(请参阅下载页面)。该项目文件包含3个个体(三个)的基因分型数据,由 Illumina HumanHap550 SNP 基因分型芯片进行基因分型。(我们假设基因型数据已经适当地聚类,信号强度值已经由 BeadStudio 软件计算出来; 如果没有,在做任何数据导出之前,加载适当的 *.EGT集群文件用于聚类)。
现在解压缩 ZIP 文件,我们将看到一个名为"tutorial"的目录。输入此目录,然后双击项目文件 tutorial.bsc。应该自动调用 BeadStudio 软件,并将基因分型数据载入软件界面:
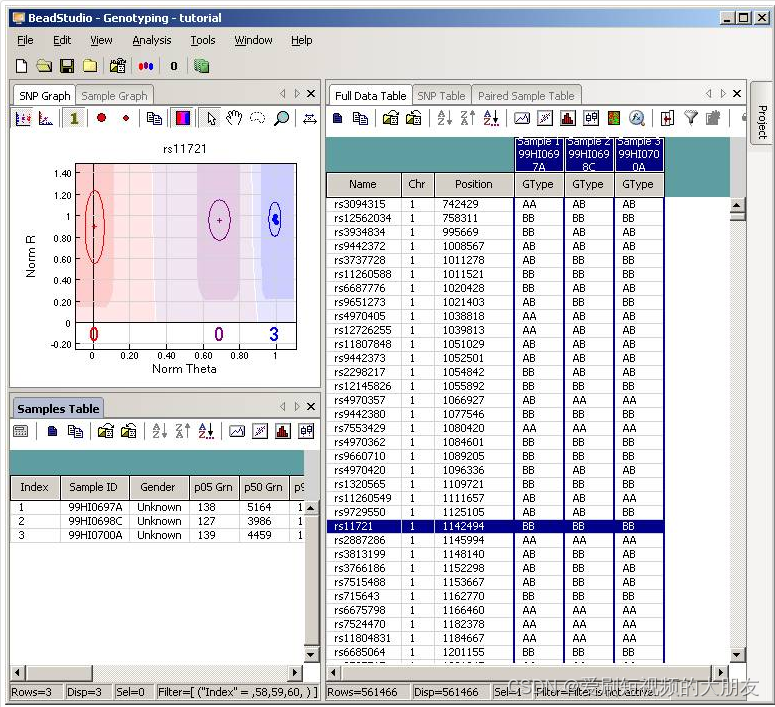 单击工具栏中的
单击工具栏中的 Column Choose 按钮(右边第三个按钮) ,将出现列选择器窗口。然后选择要包含在输出文件中的所需列(GType, Log R Ratio and B Allele Frequency)。例如,如果您看到 B Allele Freq 显示在Hidden Subcolumns框中,您可以选择它,然后单击 <=Show 按钮,这样就可以将其移动到“Displayed Subcolumns”框中。“显示的列”框至少应包含“名称”字段和所有单个标识符。强烈建议在这里也包含 Chr 和 Position 字段。您可以隐藏Address和Index之类的内容,并将它们移动到“Hidden Columns”框中。
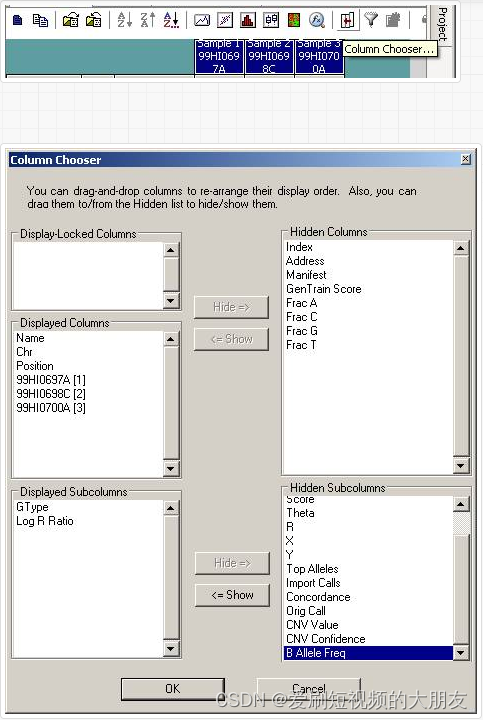
现在 BeadStudio 窗口如下所示。单击 Select All Rows 按钮(工具栏中的第一个按钮)选择所有数据:
 然后单击
然后单击 Export Displayed Data to File 按钮(工具栏中的第三个按钮) ,将该文件保存为类似 tutorial.txt 的内容。这个文件将是一个制表符分隔的文件,每行有一个 SNP,每行包含所有个体的基因型、 log R 比率和 b 等位基因频率信息。(如果您正在使用 BeadStudio 版本3,将出现一个对话框,询问您是否要导出所有数据,单击 Yes 或 No 都可以,因为您已经选择了所有数据)。Txt 的文件大小对于三个人约为50MB。
提示: 当您的项目文件包含许多(> 1000)示例时,数据导出可能非常缓慢(为了查看导出过程有多快,您可以监视输出文件大小并查看其增长速度,或者您可以使用 Windows 任务管理器查看 CPU 使用情况: 如果 beadstudio.exe 的 CPU 使用率低于10% ,那么您就有问题)。为了加快导出过程,可以使用列选择器将 Index 设置为Displayed Columns,然后单击“Sort to sort by index”,然后按列选择器隐藏 Index 列,然后再次导出数据。当数据按索引排序时,对于大样本量,导出速度要快得多; 然而,在这种情况下,标记位置在输出文件中没有按顺序排序,可能不便于后续分析。输出文件中标记的顺序不影响 PennCNV 调用 CNV。
 现在,将
现在,将 tutorial.txt 文件传输到安装了 PennCNV 的机器上,我们将使用 PennCNV 软件为这三个人生成 CNV 呼叫。
在下一步中,我们将处理包含基因型数据的 Tutorial.txt 文件,并将该文件分割为单独的文件。Txt 文件可以从 BeadStudio 软件导出(如步骤1所示) ,也可以直接从 PennCNV 网站下载页面下载(当用户跳过步骤1时)。
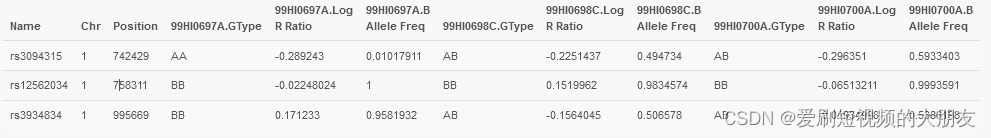 第一行称为标题行,其中包含关于每列含义的信息。后面的每行包含所有个体的每行一个 SNP 的信息。
第一行称为标题行,其中包含关于每列含义的信息。后面的每行包含所有个体的每行一个 SNP 的信息。
注意: 标题行告诉 PennCNV 每一列的含义。实际上,只有 Name 列、.Log R Ratio和.B Allele Frequency列是产生 PennCNV 的 CNV 呼叫所必需的。但是,其他列有时可能提供附加信息。例如,Chr 和 Position 列可以帮助对信号强度文件(如果尚未排序)进行排序,并检查特定区域的 LRR 和 BAF 值。*.GType 列可以帮助解释 CNV 调用并增加调用的置信度,例如推断从头 CNV 的亲本起源,或用于确认一个区域中具有多个 NC 基因型的纯合缺失。
我们需要将该文件分成几个文件,每个文件对应一个文件。我们可以使用一个标准的 Linux 命令cut来做到这一点:
[kai@adenine tutorial1 ENS40]$ cut -f 1-6 tutorial.txt > sample1.txt
[kai@adenine tutorial1 ENS40]$ cut -f 1-3,7-9 tutorial.txt > sample2.txt
[kai@adenine tutorial1 ENS40]$ cut -f 1-3,10-12 tutorial.txt > sample3.txt
上面的命令获取 tuorial.txt 文件中所需的制表符分隔的列,并为每个示例生成一个新的6列文件。Txt 文件中的前三列(Name、 Chr、 Position)保存在所有输出文件中。
或者,如果输入文件非常大(例如,包含600个个体的基因分型信息的6GB 文件) ,则由于扫描硬盘驱动器的开销(例如,扫描3.6 TB 的数据) ,多次运行 cut 命令非常缓慢。不使用 cut 程序,我们可以使用 PennCNV 包中的 kcolumn.pl 程序来实现同样的目标,但只扫描一次 inputfile 文件:
[kai@adenine tutorial1 ENS40]$ kcolumn.pl tutorial.txt split 3 -heading 3 -tab -out sample
上面的命令指定我们希望按制表符分隔的列拆分 tutorial.txt 文件,前3列是标题列(保存在每个输出文件中的列) ,每3列将写入一个不同的输出文件。tab 参数告诉程序 tutorial.txt 文件是用 tab 分隔的(默认情况下,kcolumn.pl 程序使用空格和 tab 字符来定义列) ,而--out 参数指定输出文件名应该以 sample 开头。注意,可以在命令行中同时使用双破折号或单破折号(因此--output 的含义与-output 相同) ,并且只要与另一个参数没有歧义,就可以省略参数的尾随字母(因此--output 参数与-o 或-ou 或-out 或-outp 或-outpu参数的含义相同)。还要注意,如果输入文件非常大(例如,一个包含1200个人的基因分型信息的12GB 文件) ,我们可能需要使用--start_split 和--end_split 参数并多次运行 kcolumn.pl 程序,以克服操作系统对同时处理文件的限制。例如,可以运行该程序两次: 第一次使用 -start 1 -end 1000,第二次使用-start 1001 -end 2000。
默认情况下,输出文件名将由 split1、 split2等附加; 但是,您可以使用-name_by_header 参数(或者简单的-name 参数) ,以便根据 tutorial.txt 文件第一行中的名称注释生成输出文件名。
提示:-name 参数告诉程序输出文件名应该基于输入文件第一行中的第一个单词(例如,99HI0697A 和99HI0698C)。有时,示例名称包含非单词字符,如“-”; 在这种情况下,还可以添加“--beforestring .GType”。除了-name 参数之外,还有上面命令的“ GType”参数; 这意味着应使用“ .GType”作为输出文件名,使输出文件名为 sample.99HI0697A, sample.99HI0698C 等。
要获得 kcolumn.pl 程序(或 PennCNV 包中的任何其他程序)的每个参数的更详细描述,请尝试--help 参数。要阅读 kcolumn.pl 程序(或 PennCNV 包中的任何其他程序)的完整手册,请使用--man 参数。
现在我们有三个文件,分别名为 sample1.txt、 sample2.txt 和 sample3.txt,分别对应于三个个体,我们需要为它们识别 CNV。
注意: 文件分割后,检查输出文件非常重要。通常情况下,如果在程序完成后保持终端打开,应该有一行说明所有分割操作都已完成,以确认此步骤完全成功。有时,由于缺乏硬盘空间,或者由于程序在完成之前中断,文件分割没有成功完成,导致标记数比应该包含的少。您可以执行一个简单的“ wc-l file.split1”来检查随机输出文件中的行数: 对于 Affy 6,900K,应该是大约180万行; 对于 Illumina 1M,应该是1 millino; 对于 Illumina 550K 阵列,应该是550K。如果没有,那么文件分割就不完全正确,您可以使用“ tail file.split1”查看文件中的最后一行是什么,通常该行不完整,这意味着那里有问题。再次重新进行分割,并确保在调用 CNV 之前完成文件分割。
等位基因群体频率文件
该文件提供每个标记的 PFB 信息,并将染色体坐标信息提供给 PennCNV。它是一个由制表符分隔的文本文件,有四列,表示标记名称、染色体、位置和 PFB 值。当 PFB 值为2时,这意味着该标记是一个没有多态性的 CN 标记。下面是一个例子:
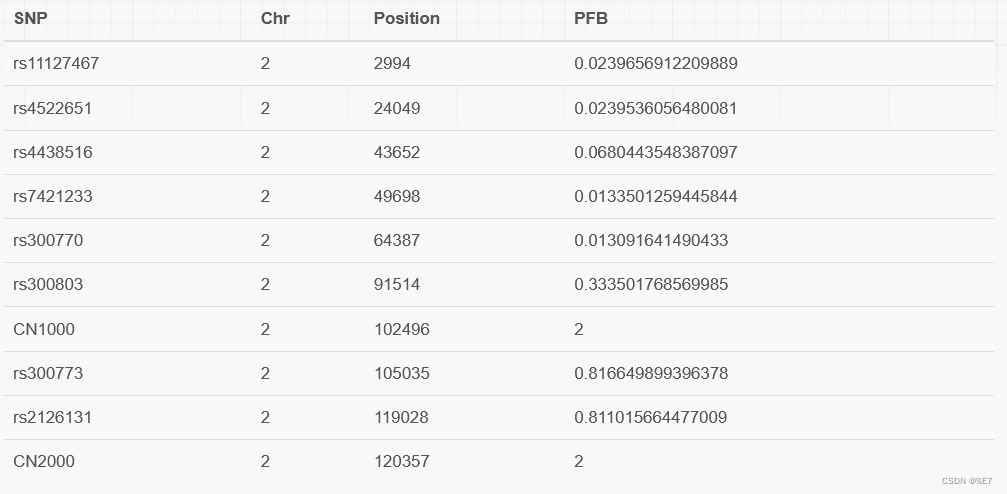 请注意,不同的阵列制造商(如 Affymetrix 和 Illumina)可能使用不同的定义 B 等位基因,即使对于相同的 SNP。
请注意,不同的阵列制造商(如 Affymetrix 和 Illumina)可能使用不同的定义 B 等位基因,即使对于相同的 SNP。
当读取信号强度文件时,PennCNV 将只处理 PFB 文件中注释的标记。因此,如果我们想从 CNV 分析中删除一些标记,由于它们的质量低,已知的问题(在节段重复区域内或在假常染色体区域内) ,我们可以简单地从 PFB 文件中删除这些标记,而不改变信号强度文件本身。类似地,如果我们想在不同的基因组组装上调用 CNV (NCBI36与 NCBI35) ,我们可以简单地更改 PFB 文件以反映新的染色体坐标,而不需要更改信号强度文件。
HMM 文件
HMM 文件为 PennCNV 提供了 HMM 模型,并告诉程序不同拷贝数状态的预期信号强度值,以及不同拷贝数状态的预期转换概率。下面是一个例子:
 第一个大块(A)指定转换概率。然而,在 PennCNV 中,转移概率取决于相邻标记之间的距离。因此,这里的数字是5kb 距离的平均数字,实际的转移概率是使用前面描述的公式计算的,该公式根据距离进行调整。
第一个大块(A)指定转换概率。然而,在 PennCNV 中,转移概率取决于相邻标记之间的距离。因此,这里的数字是5kb 距离的平均数字,实际的转移概率是使用前面描述的公式计算的,该公式根据距离进行调整。
第二个大块(B)不被 PennCNV 使用(非常原始的 PennCNV 程序改编自使用这种 HMM 模型文件格式的 UMDHMM 框架,因此为了与以前版本的兼容性,在所有更新版本中使用相同的 HMM 格式,尽管事实上许多字段都是无用的)。Pi 值指定 HMM 链中不同拷贝数状态的初始概率。
第三个大块(B1,B2,B3)指定预期(平均)信号强度值,以及它们的标准差。B1用于 LRR,B2用于 BAF,而 B3用于 CN 标记的 LRR,这些标记通常比 SNP 标记具有更大的方差。两个信号强度值,包括 LRR 和 BAF,都被 PennCNV 使用。此外,PennCNV 使用 UF 因子来指定预期坏标记的分数,即表现不好且不符合预期的标记。对于 Illumina 阵列,0.01似乎是一个不错的选择; 对于 Affymetrix 阵列,这个值应该略高于0.03或0.05。显然,这个数值反映了调用算法的灵敏度,尤其是在非常小的 CNV 区域: 当使用较高的 UF 值时,来自较小但实际 CNV 区域的信号可以被视为数据中的随机噪声,但该算法对信号值的随机波动更具鲁棒性)。
GCModel 文件
该文件指定每个标记周围1Mb 基因组区域(每边500kb)的 GC 内容。它被 PennCNV 中的-gcmodel 论证所使用,并且已经被用于挽救受基因组波影响的样品(详见 Diskin 等人)。下面是一个例子:
请注意,PennCNV 没有使用第二和第三列,因为这些信息已经在 PFB 文件中提供了。GC 值范围从0到100,表示标记周围每个区域中 G 或 C 碱基对的百分比。
List file
虽然信号文件名可以在命令行中提供,但是 PennCNV 中的-list 参数可以获取一个列表文件,该列表文件提供要处理的所有文件名。
当为每个人调用 CNV 时,列表文件应该每行包含一个文件名。当为 trios 调用 CNV 时(使用-three 参数或-join 参数) ,列表文件应该每行包含三个由制表符分隔的文件名。当为四进制调用 CNV 时,列表文件每行包含四个文件名,以制表符分隔。
如果列表文件中的文件名被’字符包围,它将被视为系统命令,而不是文件名。PennCNV将执行这个系统命令,从这个命令中读取信号值,然后生成CNV调用。一个明显的例子是将信号值存储在压缩文件中(例如sample1.txt.gz),然后使用gunzip -c sampl1.txt.gz作为PennCNV的输入文件名。另外,一些中心可能将所有信号强度值存储在关系数据库中,并编写自定义脚本来检索这些信号值并以制表符分隔的格式输出。在这种情况下,可以按照上述方式在列表文件中指定这些自定义脚本,以供PennCNV直接处理。
这篇关于CNV学习3(CNV软件使用学习--input)的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!





