本文主要是介绍iMeta文献观点:16s全长-功能预测更具优势,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
二代扩增子测序技术由于其读长短,信息量有限,在微生物组功能预测领域的结果一直备受争议。标记基因的分类学分辨率直接关系到微生物组功能预测的准确性及可信度,那么,基于二代扩增子测序结果的功能预测是否可信,有没有更好的方法可以实现更高效精准的功能预测研究。
近日,巴西圣保罗医院肿瘤分子诊断中心Vitor Heidrich教授和德国朱利叶斯库恩研究所Lukas Beule教授在iMeta在线发表文章,深入探讨了功能预测研究的准确性问题,提出了利用分类组成(特别是来源于短读长测序时)预测微生物组功能之生物学可信度方面的一些担忧,讨论了标记基因的分类学分辨率、标记基因的基因组内变异以及微生物组数据的组成性质,并指出16S全长测序将逐步取代短读长测序,成为预测微生物组功能的主力。

发表期刊:iMeta
发表时间:2022. 07
DOI: 10.1002/imt2.38
文章亮点
本研究阐述了利用微生物组数据集分类谱预测微生物潜在功能之生物学可信度方面的一些问题
讨论了标记基因的分类学分辨率、标记基因的基因组内变异以及微生物组数据的组成性质
将微生物组功能的实际测定与预测相结合是理解微生物组功能的一个好办法
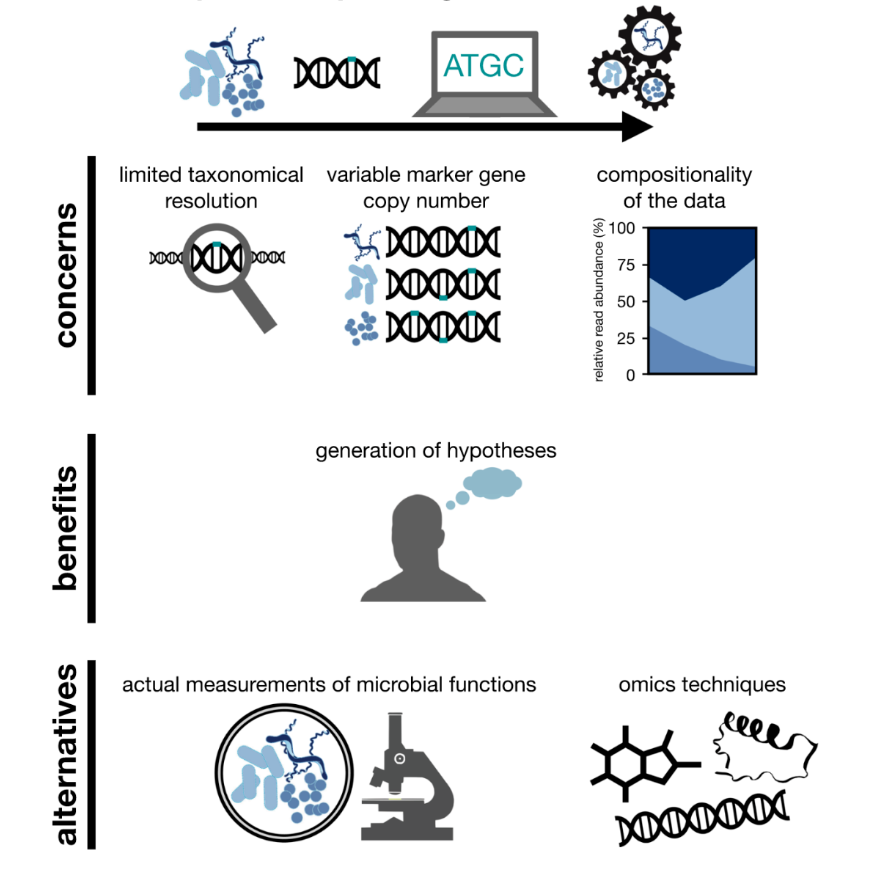
图1 从扩增子测序微生物组数据集预测微生物组功能潜力的关注点、好处和替代方案的示意图
主要内容
1、扩增子测序的分类学分辨率
使用Illumina平台的短读长扩增子测序是目前微生态研究中最常见的测序技术。PE300测序模式可获得2x300bp的碱基对信息。在微生物分析中,短读长扩增子测序最常用的对象是具有分类学信息的细菌16S核糖体RNA(rRNA)和真菌内转录间隔子(ITS)基因位点,但对这些基因基于illumina平台进行部分区域测序往往在种水平的区分度不高,且大多无法区分相关度高的菌株。
Loop Genomics提出可通过采用Illumina短读长测序技术,结合在每个16S扩增子独特的标签序列来合成长读长序列等方法,可以提高分类学水平。但这些替代方法不是最优选择,因为与Loop Genomics技术相反,全长16S序列无法用这种策略重建。
已有研究证明,采用PacBio等长读长测序技术可以实现16S全长测序,提高分类学分辨率。16S全长测序可针对16S rRNA全长9个可变区进行测序,比单一区段能提供更多信息,真正精确到“种”的分类鉴定,同时可准确识别序列多态性,定位菌株水平的变异,检测的灵敏度更高,能够挖掘新物种,更加真实的还原样本微生物群落结构。
尽管16S全长测序广泛应用的成本仍然高昂,但从长远来看,通过长读长扩增子测序或基于唯一识别的短读长扩增子测序的生物信息学重建实现的16S全长测序将逐渐取代短读长测序,提高了分类学分辨率,进而更准确地预测微生物的潜在功能。
2、标记基因的基因组内变异性
利用扩增子测序数据分析微生物组结构的另一个难点是微生物基因组中标记基因的拷贝数有差异,这种变异性有时会认为是等位基因多样性,甚至会混淆微生物组的组成。同时,标记基因的基因组内变异性会干扰潜在功能的相对重要性估计,进而可能会高估潜在功能谱的多样性。NGS测序由于其读长短,测序范围有限,无法准确鉴定基因组内的基因变异性,而16S全长测序覆盖度和准确度更高,可以准确识别序列多态性,定位菌株水平的变异。
3、微生物组数据的组成性质
由于NGS从样本中产生的序列读数与样本中的细菌细胞数量之间没有关系,因此这个读数并不转化为细菌丰度。NGS中每个分类单元产生的读数仅反映了群落一部分的相对大小,使NGS微生物组数据集是可组装的。换句话说,这意味着它解锁了一个微生物群落中类群的相对测序读取丰度(即比例或频率),但由于整个群落的大小(微生物生物量)仍然未知,它没能揭示类群的绝对丰度。
因此,即使一个群落的预测功能谱与其实际功能相吻合,但由于未考虑该群落的总种群规模,而无法对功能潜力的大小进行估计。目前也有很多其他的微生物定量方法可以克服微生物组分析的这一局限性,但是不同的量化方法可能会引入额外的数据异质性。
另一种规避微生物组成数据分析问题的策略是使用比率,因为使用比率可以消除微生物自身负荷造成的偏差。此外,PCR扩增偏差是一个众所周知的误差来源,它可以曲解群落组成和功能预测。
4、测定微生物群的实际功能
另一种要求更高的方法是在尽量测定微生物群的实际功能。但是,只有当微生物群落可获取,在采样时相应的功能是活跃的,并且有足够的种群大小和采样量用于检测,测量微生物的实际功能才是可行的。对于不满足这些标准的微生物群,使用实时荧光定量PCR等额外技术是对某类功能基因遗传潜能定量表征的一种有意义的补充。但必须指出的是,这些遗传潜能并不一定转化为微生物活动和过程。
从这个角度来说,转录组学、蛋白质组学和代谢组学等组学技术有助于探索已经表达的遗传潜能。结合功能预测和特定功能的实际测定是了解微生物功能的一个强有力的方法。
5、产生和验证假设
尽管有局限性,但本研究承认功能预测有产生新假设的特殊潜力。然而,尤其不能忽视微生物群落是复杂的。因此,功能组成是高维的,很难分析。这意味着通常潜在功能预测工具会引发太多的研究方向,而产生直接的假设变得困难。本研究强烈鼓励研究人员选择有意义的假设,并尽可能独立地检验这些假设,例如代谢组、实时荧光定量PCR等来进行验证。
总结
总体而言,本研究肯定那些为实现微生物组分类学数据集功能预测所做的努力,也坚信功能预测有助于产生新的思路和潜在研究方向。但是,本研究同时认为在提取微生物组DNA的基础上进行短读长扩增子测序预测微生物组功能不应作为生物学推断的唯一基准,而从短读到长读长测序技术的转变将有助于克服这些问题。此外,在功能预测的同时,结合使用组学(如代谢组学)和非组学方法(如qPCR)表征微生物活性对阐明微生物组功能至关重要。
参考文献:
Are short‐read amplicons suitable for the prediction ofmicrobiome functional potential? A critical perspective. iMeta 1: e38, 2022.
DOI: 10.1002/imt2.38
这篇关于iMeta文献观点:16s全长-功能预测更具优势的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!





