本文主要是介绍PSP - AlphaFold2 的 2.3.2 版本源码解析 (1),希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
欢迎关注我的CSDN:https://spike.blog.csdn.net/
本文地址:https://blog.csdn.net/caroline_wendy/article/details/130323566
时间:2023.4.22
官网:https://github.com/deepmind/alphafold
AlphaFold2是一种基于深度学习的方法,根据氨基酸序列预测蛋白质的三维结构。在2020年的CASP14竞赛中取得了突破性的成绩,达到了原子级的精度。AlphaFold2的核心是Evoformer模块,利用多序列比对(MSA)和残基对特征,来捕捉蛋白质的共进化信息和物理约束。Evoformer的输出被送入一个结构模块,使用注意力机制和几何约束来生成蛋白质的骨架和侧链坐标。AlphaFold2的源代码和预训练模型已经公开发布,为蛋白质结构预测领域带来了新的机遇和挑战。
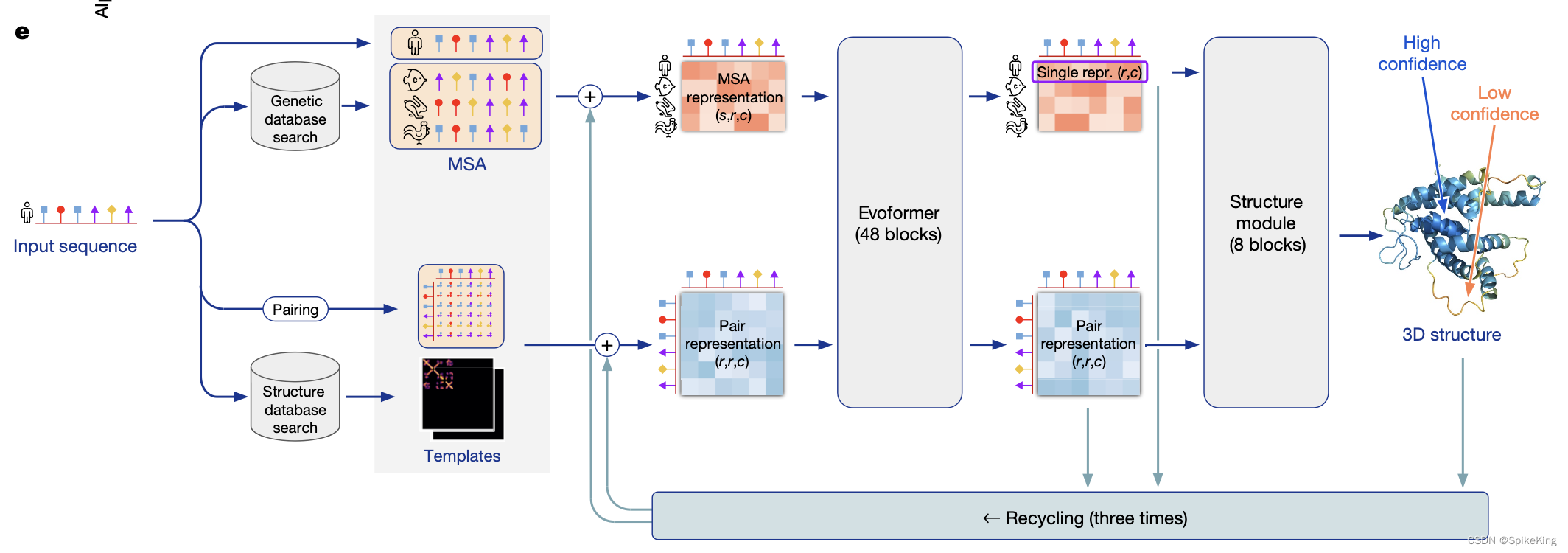
AlphaFold版本2.3.2+:
commit 4d83e3fc0883011e3d597eb6d33e532267754708 (HEAD, origin/main, origin/HEAD)
Author: Hamish Tomlinson <htomlinson@google.com>
Date: Tue Apr 11 02:22:30 2023 -0700Bump version to 2.3.2 in setup.py and colab.PiperOrigin-RevId: 523341685Change-Id: Ie332a4db056adb2d42bcd2f4006e4258b8908507commit 3f31725591b0c6b0b6d63214235d3abe6b81742c (tag: v2.3.2)
Author: Hamish Tomlinson <htomlinson@google.com>
Date: Mon Mar 27 04:50:17 2023 -0700Upgrade pyopenssl in colab.PiperOrigin-RevId: 519687065Change-Id: Ib72232f3f4d09ee8f3a2cc312d7a83d8b892a23e
输入序列117个氨基酸的序列,即CASP15的T1104序列:
>T1104 EntV136 , Enterococcus faecalis, 117 residues|
QLEDSEVEAVAKGLEEMYANGVTEDNFKNYVKNNFAQQEISSVEEELNVNISDSCVANKIKDEFFAMISISAIVKAAQKKAWKELAVTVLRFAKANGLKTNAIIVAGQLALWAVQCG
1. 入口函数
脚本:run_alphafold.py
- 单链使用
pipeline.DataPipeline()类,多链使用pipeline_multimer.DataPipeline()类,关注单链逻辑。 - 核心逻辑位于
run_alphafold.py#predict_structure()。
源码如下:
monomer_data_pipeline = pipeline.DataPipeline(jackhmmer_binary_path=FLAGS.jackhmmer_binary_path,hhblits_binary_path=FLAGS.hhblits_binary_path,uniref90_database_path=FLAGS.uniref90_database_path,mgnify_database_path=FLAGS.mgnify_database_path,bfd_database_path=FLAGS.bfd_database_path,uniref30_database_path=FLAGS.uniref30_database_path,small_bfd_database_path=FLAGS.small_bfd_database_path,template_searcher=template_searcher,template_featurizer=template_featurizer,use_small_bfd=use_small_bfd,use_precomputed_msas=FLAGS.use_precomputed_msas)
...
data_pipeline = monomer_data_pipeline
...
for i, fasta_path in enumerate(FLAGS.fasta_paths):fasta_name = fasta_names[i]predict_structure(fasta_path=fasta_path,fasta_name=fasta_name,output_dir_base=FLAGS.output_dir,data_pipeline=data_pipeline,model_runners=model_runners,amber_relaxer=amber_relaxer,benchmark=FLAGS.benchmark,random_seed=random_seed,models_to_relax=FLAGS.models_to_relax)
在predict_structure()中,AF2完成全部的结构预测过程,核心逻辑位于data_pipeline.process(),输入fasta文件和MSA路径,位于alphafold/data/pipeline.py#process()。
源码如下:
def predict_structure(fasta_path: str,fasta_name: str,output_dir_base: str,data_pipeline: Union[pipeline.DataPipeline, pipeline_multimer.DataPipeline],model_runners: Dict[str, model.RunModel],amber_relaxer: relax.AmberRelaxation,benchmark: bool,random_seed: int,models_to_relax: ModelsToRelax):"""Predicts structure using AlphaFold for the given sequence."""...feature_dict = data_pipeline.process(input_fasta_path=fasta_path,msa_output_dir=msa_output_dir)...
在alphafold/data/pipeline.py#process()中,核心逻辑是:
- 搜索MSA和Template,其中,MSA搜索UniRef90、BFD、MGnify三个库,Template搜索
pdb70和pdb_seqres两个文件,PDB库在RCSB的mmcif中查找。 - 初始化三类特征,之后再进行更新,即序列特征(
make_sequence_features)、MSA特征(make_msa_features)、Template特征(get_templates)。
源码如下:
def process(self, input_fasta_path: str, msa_output_dir: str) -> FeatureDict:"""Runs alignment tools on the input sequence and creates features."""with open(input_fasta_path) as f:input_fasta_str = f.read()input_seqs, input_descs = parsers.parse_fasta(input_fasta_str)if len(input_seqs) != 1:raise ValueError(f'More than one input sequence found in {input_fasta_path}.')input_sequence = input_seqs[0]input_description = input_descs[0]num_res = len(input_sequence)uniref90_out_path = os.path.join(msa_output_dir, 'uniref90_hits.sto')jackhmmer_uniref90_result = run_msa_tool(msa_runner=self.jackhmmer_uniref90_runner,input_fasta_path=input_fasta_path,msa_out_path=uniref90_out_path,msa_format='sto',use_precomputed_msas=self.use_precomputed_msas,max_sto_sequences=self.uniref_max_hits)mgnify_out_path = os.path.join(msa_output_dir, 'mgnify_hits.sto')jackhmmer_mgnify_result = run_msa_tool(msa_runner=self.jackhmmer_mgnify_runner,input_fasta_path=input_fasta_path,msa_out_path=mgnify_out_path,msa_format='sto',use_precomputed_msas=self.use_precomputed_msas,max_sto_sequences=self.mgnify_max_hits)msa_for_templates = jackhmmer_uniref90_result['sto']msa_for_templates = parsers.deduplicate_stockholm_msa(msa_for_templates)msa_for_templates = parsers.remove_empty_columns_from_stockholm_msa(msa_for_templates)if self.template_searcher.input_format == 'sto':pdb_templates_result = self.template_searcher.query(msa_for_templates)elif self.template_searcher.input_format == 'a3m':uniref90_msa_as_a3m = parsers.convert_stockholm_to_a3m(msa_for_templates)pdb_templates_result = self.template_searcher.query(uniref90_msa_as_a3m)else:raise ValueError('Unrecognized template input format: 'f'{self.template_searcher.input_format}')pdb_hits_out_path = os.path.join(msa_output_dir, f'pdb_hits.{self.template_searcher.output_format}')with open(pdb_hits_out_path, 'w') as f:f.write(pdb_templates_result)uniref90_msa = parsers.parse_stockholm(jackhmmer_uniref90_result['sto'])mgnify_msa = parsers.parse_stockholm(jackhmmer_mgnify_result['sto'])pdb_template_hits = self.template_searcher.get_template_hits(output_string=pdb_templates_result, input_sequence=input_sequence)if self._use_small_bfd:bfd_out_path = os.path.join(msa_output_dir, 'small_bfd_hits.sto')jackhmmer_small_bfd_result = run_msa_tool(msa_runner=self.jackhmmer_small_bfd_runner,input_fasta_path=input_fasta_path,msa_out_path=bfd_out_path,msa_format='sto',use_precomputed_msas=self.use_precomputed_msas)bfd_msa = parsers.parse_stockholm(jackhmmer_small_bfd_result['sto'])else:bfd_out_path = os.path.join(msa_output_dir, 'bfd_uniref_hits.a3m')hhblits_bfd_uniref_result = run_msa_tool(msa_runner=self.hhblits_bfd_uniref_runner,input_fasta_path=input_fasta_path,msa_out_path=bfd_out_path,msa_format='a3m',use_precomputed_msas=self.use_precomputed_msas)bfd_msa = parsers.parse_a3m(hhblits_bfd_uniref_result['a3m'])templates_result = self.template_featurizer.get_templates(query_sequence=input_sequence,hits=pdb_template_hits)sequence_features = make_sequence_features(sequence=input_sequence,description=input_description,num_res=num_res)msa_features = make_msa_features((uniref90_msa, bfd_msa, mgnify_msa))logging.info('Uniref90 MSA size: %d sequences.', len(uniref90_msa))logging.info('BFD MSA size: %d sequences.', len(bfd_msa))logging.info('MGnify MSA size: %d sequences.', len(mgnify_msa))logging.info('Final (deduplicated) MSA size: %d sequences.',msa_features['num_alignments'][0])logging.info('Total number of templates (NB: this can include bad ''templates and is later filtered to top 4): %d.',templates_result.features['template_domain_names'].shape[0])return {**sequence_features, **msa_features, **templates_result.features}
2. 初始化序列特征
核心逻辑alphafold/data/pipeline.py#make_sequence_features(),逻辑简单:
- 输入:序列、描述、序列长度(残基个数)
- 输出:
aatype(氨基酸类型)、between_segment_residues(片段残基之间)、domain_name(序列描述)、residue_index(残基索引)、seq_length(序列长度)、sequence(序列)
一般而言,在CASP15中,序列都是一个域(domain),因此,序列描述使用 domain_name 表示。
调用过程,源码如下:
input_sequence = input_seqs[0]
input_description = input_descs[0]
num_res = len(input_sequence)sequence_features = make_sequence_features(sequence=input_sequence,description=input_description,num_res=num_res)
生成序列特征,源码如下:
def make_sequence_features(sequence: str, description: str, num_res: int) -> FeatureDict:"""Constructs a feature dict of sequence features."""features = {}features['aatype'] = residue_constants.sequence_to_onehot(sequence=sequence,mapping=residue_constants.restype_order_with_x,map_unknown_to_x=True)features['between_segment_residues'] = np.zeros((num_res,), dtype=np.int32)features['domain_name'] = np.array([description.encode('utf-8')],dtype=np.object_)features['residue_index'] = np.array(range(num_res), dtype=np.int32)features['seq_length'] = np.array([num_res] * num_res, dtype=np.int32)features['sequence'] = np.array([sequence.encode('utf-8')], dtype=np.object_)return features
在序列特征中,以序列长度117为例,包括6类特征,具体输出,如下:
aatype(氨基酸类型):(117, 21),数据one-hot类型,21维,即20个残基 + 1个X未知,其中,117是序列长度。between_segment_residues(片段残基之间):(117,),全0列表。domain_name(序列描述):(1,),FASTA的描述。
array([b'T1104 EntV136 , Enterococcus faecalis, 117 residues|'], dtype=object)
residue_index(残基索引):(117,),残基索引,0-116的数字排列。
array([ 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12,13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25,26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38,39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51,52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64,65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77,78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90,91, 92, 93, 94, 95, 96, 97, 98, 99, 100, 101, 102, 103,104, 105, 106, 107, 108, 109, 110, 111, 112, 113, 114, 115, 116],dtype=int32)
seq_length(序列长度):(117,),序列长度,数值都是117。sequence(序列):即输入序列。
array([b'QLEDSEVEAVAKGLEEMYANGVTEDNFKNYVKNNFAQQEISSVEEELNVNISDSCVANKIKDEFFAMISISAIVKAAQKKAWKELAVTVLRFAKANGLKTNAIIVAGQLALWAVQCG'], dtype=object)
3. 初始化MSA特征
核心逻辑alphafold/data/pipeline.py#make_msa_features(),调用过程:
msa_features = make_msa_features((uniref90_msa, bfd_msa, mgnify_msa))
3.1 搜索和解析MSA
make_msa_features函数,输入包括3个不同库的MSA文件,即UniRef90、MGnify、BFD,其中BFD可选规模的大小。
- UniRef90:包含1.3亿条氨基酸序列的数据库,通过将 UniProtKB 和其他来源的序列按照 90% 的相似度聚类而生成的,占用空间约为30GB。
- MGnify (MetaGenomics Unify) :包含10亿条氨基酸序列的数据库,属于微生物宏基因组学(MetaGenomics),来源于环境样本中DNA的测序和分析,占用空间约为100GB。
- BFD (Big Fantastic Database) :包含25亿条氨基酸序列的数据库,通过对多种来源的蛋白质结构和序列进行整合而生成的,占用空间约为2.5TB。
源码如下:
uniref90_msa = parsers.parse_stockholm(jackhmmer_uniref90_result['sto'])
mgnify_msa = parsers.parse_stockholm(jackhmmer_mgnify_result['sto'])
if self._use_small_bfd:bfd_msa = parsers.parse_stockholm(jackhmmer_small_bfd_result['sto'])
else:bfd_msa = parsers.parse_a3m(hhblits_bfd_uniref_result['a3m'])
其中,UniRef90和MGnify都是使用jackhmmer进行搜索,而大规模BFD使用hhblits,小规模BFD使用jackhmmer。
对于搜索出的MSA序列,再进行解析和清洗,保持格式一致,即去除没有对齐的残基,保证序列长度与输入序列一致,同时,保留删除的位置(deletion_matrix)。参考alphafold/data/parsers.py#parse_a3m()&parse_stockholm(),源码如下:
# parse_a3m()return Msa(sequences=aligned_sequences,deletion_matrix=deletion_matrix,descriptions=descriptions)# parse_stockholm()return Msa(sequences=msa,deletion_matrix=deletion_matrix,descriptions=list(name_to_sequence.keys()))
其中,输出的MSA,包括2类格式,即a3m和sto。a3m格式,第1个是输入序列,其他是搜索序列,需要删除小写字母才能与输入序列保持一致,如下:
>T1104 EntV136 , Enterococcus faecalis, 117 residues|
QLEDSEVEAVAKGLEEMYANGVTEDNFKNYVKNNFAQQEISSVEEELNVNISDSCVANKIKDEFFAMISISAIVKAAQKKAWKELAVTVLRFAKANGLKTNAIIVAGQLALWAVQCG
>Q6DRR6 107 0.440 9.748E-24 1 116 117 42 175 179
-IEEQrQIDEVAAVLEKMFADGVTEENLKQYAQANYSEEELIIADNELNTNlsqiqdenaimykvDWgalGNCMANKIKDELLAMISVGTIIKYAQKKAWKELAKIVIKYVAKAGVKTNAALIAGQLAIWGLQCG
>U6S4W9 107 0.871 9.748E-24 0 116 117 57 188 191
QLEDSEVEAVAKGLEEMYANGVTEDNFKNYVKNNFAQQEISSVEEELNVNIsdastvvqarfnWNalgSCVANKIKDEFFAMISISAIVKAAQKKAWKELAVTVLRFAKANGLKTNAIIVAGQLALWAVQCG
sto格式,sto即Stockholm,多序列比对(MSA)格式,第一块是查询结果,第二块是所有匹配目标序列的比对结果,如下:
# STOCKHOLM 1.0
#=GF ID 7c5664d51e2f56827f5232132517161a-i1
#=GF AU jackhmmer (HMMER 3.3.2)#=GS tr|A0A1X3AJN2|A0A1X3AJN2_ENTFL/18-73 DE [subseq from] Streptococcin A-M57 OS=Enterococcus faecalis OX=1351 GN=ELS84_1737 PE=4 SV=1
#=GS tr|A0A1X3AJN2|A0A1X3AJN2_ENTFL/75-149 DE [subseq from] Streptococcin A-M57 OS=Enterococcus faecalis OX=1351 GN=ELS84_1737 PE=4 SV=1
...
T1104 EntV136 , Enterococcus faecalis, 117 residues| QLEDSEVEAVAKGLEEMYANGV--TEDNF---K---NYVKNNFAQQEIS---------------S-----VEEELNV--------NISD-----------S-------------CVANKIKDEFFAMISISAIVKAAQKKAWKELAVTVLRFAKANGLKTNAIIVAGQLALWAVQCG
tr|A0A1X3AJN2|A0A1X3AJN2_ENTFL/18-73 QLEDSEVEAVAKGLEEMYANGV--TEDNF---K---NYVKNNFAQQEIS---------------S-----VEEELNV--------NISD-----------A-------------ST-------------------------------------------------------------
#=GR tr|A0A1X3AJN2|A0A1X3AJN2_ENTFL/18-73 PP 89********************..*****...*...*************...............*.....*******........****...........8.............64.............................................................
tr|A0A1X3AJN2|A0A1X3AJN2_ENTFL/75-149 ----------------------------------------------------------------------VQARFNW--------NALG-----------S-------------CVANKIKDEFFAMISISAIVKAAQKKAWKELAVTVLRFAKANGLKTNAIIVAGQLALWAVQCG
#=GR tr|A0A1X3AJN2|A0A1X3AJN2_ENTFL/75-149 PP ......................................................................6678899........99**...........*.............**************************************************************9
3.2 计算MSA特征
核心逻辑alphafold/data/pipeline.py#make_msa_features():
- 通过集合去重,即
seen_sequences = set(); - 输出特征。
在MSA特征中,以序列长度117、MSA数量151为例,包括4类特征,具体输出如下:
deletion_matrix_int,因序列对齐所删除的残基矩阵,(151, 117),稀疏矩阵,0较多,只保留连续个数,例如:
# 序列
-----DAKEVAEQLEFIFEEAAIKDiNDKiigldiEMIEEKYGPGaELQQLKEEMN---------------------------------------------------------------------
# deletion vec
[0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 1, 0, 0, 6, 0, 0, 0, 0, 0, 0, 0, 0, 0, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0]
msa,残基索引矩阵,(151, 117),例如:
array([[21, 21, 3, ..., 13, 1, 5],[13, 9, 3, ..., 13, 1, 5],[13, 7, 8, ..., 13, 1, 5],...,[21, 21, 21, ..., 18, 1, 21],[21, 21, 21, ..., 21, 21, 21],[21, 21, 21, ..., 21, 21, 21]], dtype=int32)
num_alignments,MSA的序列数量矩阵,例如:
array([151, 151, 151, 151, 151, 151, 151, 151, 151, 151, 151, 151, 151,151, 151, 151, 151, 151, 151, 151, 151, 151, 151, 151, 151, 151,151, 151, 151, 151, 151, 151, 151, 151, 151, 151, 151, 151, 151,151, 151, 151, 151, 151, 151, 151, 151, 151, 151, 151, 151, 151,151, 151, 151, 151, 151, 151, 151, 151, 151, 151, 151, 151, 151,151, 151, 151, 151, 151, 151, 151, 151, 151, 151, 151, 151, 151,151, 151, 151, 151, 151, 151, 151, 151, 151, 151, 151, 151, 151,151, 151, 151, 151, 151, 151, 151, 151, 151, 151, 151, 151, 151,151, 151, 151, 151, 151, 151, 151, 151, 151, 151, 151, 151, 151],dtype=int32)
msa_species_identifiers,序列物种标识,sto格式的具备物种标识,通过正则获取,例如:tr|A0A146SKV9|A0A146SKV9_FUNHE。- 参考:
alphafold/data/msa_identifiers.py#_parse_sequence_identifier()
- 参考:
源码:
def make_msa_features(msas: Sequence[parsers.Msa]) -> FeatureDict:"""Constructs a feature dict of MSA features."""if not msas:raise ValueError('At least one MSA must be provided.')int_msa = []deletion_matrix = []species_ids = []seen_sequences = set()for msa_index, msa in enumerate(msas):if not msa:raise ValueError(f'MSA {msa_index} must contain at least one sequence.')for sequence_index, sequence in enumerate(msa.sequences):if sequence in seen_sequences:continueseen_sequences.add(sequence)int_msa.append([residue_constants.HHBLITS_AA_TO_ID[res] for res in sequence])deletion_matrix.append(msa.deletion_matrix[sequence_index])identifiers = msa_identifiers.get_identifiers(msa.descriptions[sequence_index])species_ids.append(identifiers.species_id.encode('utf-8'))num_res = len(msas[0].sequences[0])num_alignments = len(int_msa)features = {}features['deletion_matrix_int'] = np.array(deletion_matrix, dtype=np.int32)features['msa'] = np.array(int_msa, dtype=np.int32)features['num_alignments'] = np.array([num_alignments] * num_res, dtype=np.int32)features['msa_species_identifiers'] = np.array(species_ids, dtype=np.object_)return features
4. 初始化Template特征
调用逻辑:
- 搜索MSA,使用jackhmmer,搜索uniref90,复用MSA部分的逻辑。
- 清洗MSA,去除重复数据(
deduplicate_stockholm_msa)、删除空列(remove_empty_columns_from_stockholm_msa)。 - 搜索Template,搜索库
jackhmmer_uniref90。 template_searcher提取Template信息。template_featurizer获取Template结果。
初始化Template特征,相关的源码,如下:
# 搜索MSA
uniref90_out_path = os.path.join(msa_output_dir, 'uniref90_hits.sto')
jackhmmer_uniref90_result = run_msa_tool(msa_runner=self.jackhmmer_uniref90_runner, input_fasta_path=input_fasta_path,msa_out_path=uniref90_out_path, msa_format='sto', use_precomputed_msas=self.use_precomputed_msas, max_sto_sequences=self.uniref_max_hits)# 清洗MSA
msa_for_templates = jackhmmer_uniref90_result['sto']
msa_for_templates = parsers.deduplicate_stockholm_msa(msa_for_templates)
msa_for_templates = parsers.remove_empty_columns_from_stockholm_msa(msa_for_templates)# 搜索Template
if self.template_searcher.input_format == 'sto':pdb_templates_result = self.template_searcher.query(msa_for_templates)
elif self.template_searcher.input_format == 'a3m':uniref90_msa_as_a3m = parsers.convert_stockholm_to_a3m(msa_for_templates)pdb_templates_result = self.template_searcher.query(uniref90_msa_as_a3m)
else:raise ValueError('Unrecognized template input format: f'{self.template_searcher.input_format}')# 提取Template信息
pdb_template_hits = self.template_searcher.get_template_hits(output_string=pdb_templates_result, input_sequence=input_sequence)# 获取Template结果
templates_result = self.template_featurizer.get_templates(query_sequence=input_sequence, hits=pdb_template_hits)
4.1 清洗Template的MSA
其中,去除重复数据,deduplicate_stockholm_msa函数,具体:
- 源码:
alphafold/data/parsers.py#deduplicate_stockholm_msa()。 - 根据
query_align设置 mask 信息。 - 根据 mask 信息,压缩搜索出的 alignment,使用set进行过滤
- 提取过滤之后的行。
源码:
...
seen_sequences = set()
seqnames = set()
# First alignment is the query.
query_align = next(iter(sequence_dict.values()))
mask = [c != '-' for c in query_align] # Mask is False for insertions.
for seqname, alignment in sequence_dict.items():# Apply mask to remove all insertions from the string.masked_alignment = ''.join(itertools.compress(alignment, mask))if masked_alignment in seen_sequences:continueelse:seen_sequences.add(masked_alignment)seqnames.add(seqname)
...
搜索出101个Template,进行去除重复,由101个Template下降为34个Template。示例如下:
# 输入
QLEDSEVEAVAKGLEEMYANGV--TEDNF---K---NYVKNNFAQQEIS---------------S-----VEEELNV--------NISD-----------S-------------CVANKIKDEFFAMISISAIVKAAQKKAWKELAVTVLRFAKANGLKTNAIIVAGQLALWAVQCG
# 压缩
QLEDSEVEAVAKGLEEMYANGVTEDNFKNYVKNNFAQQEISSVEEELNVNISDSCVANKIKDEFFAMISISAIVKAAQKKAWKELAVTVLRFAKANGLKTNAIIVAGQLALWAVQCG# 输入
QLEDSEVEAVAKGLEEMYANGV--TEDNF---K---NYVKNNFAQQEIS---------------S-----VEEELNV--------NISD-----------A-------------ST-------------------------------------------------------------
# 压缩
QLEDSEVEAVAKGLEEMYANGVTEDNFKNYVKNNFAQQEISSVEEELNVNISDAST-------------------------------------------------------------
其中,删除空列,remove_empty_columns_from_stockholm_msa函数。
- 源码位于:
alphafold/data/parsers.py#remove_empty_columns_from_stockholm_msa()。 - 根据最后一列的
#=GC RF标识符进行处理,删除不需要的列,用于不同query的Template融合。
源码如下:
def remove_empty_columns_from_stockholm_msa(stockholm_msa: str) -> str:"""Removes empty columns (dashes-only) from a Stockholm MSA."""processed_lines = {}unprocessed_lines = {}for i, line in enumerate(stockholm_msa.splitlines()):if line.startswith('#=GC RF'):reference_annotation_i = ireference_annotation_line = line
...
输入34个Template,输出34个Template。示例如下:
7c5664d51e2f56827f5232132517161a QLEDSEVEAVAKGLEEMYANGV--TEDNF---K---NYVKNNFAQQEIS---------------S-----VEEELNV--------NISD-----------S-------------CVANKIKDEFFAMISISAIVKAAQKKAWKELAVTVLRFAKANGLKTNAIIVAGQLALWAVQCG
tr|A0A1X3AJN2|A0A1X3AJN2_ENTFL/18-73 QLEDSEVEAVAKGLEEMYANGV--TEDNF---K---NYVKNNFAQQEIS---------------S-----VEEELNV--------NISD-----------A-------------ST-------------------------------------------------------------
...
#=GC RF xxxxxxxxxxxxxxxxxxxxxx..xxxxx...x...xxxxxxxxxxxxx...............x.....xxxxxxx........xxxx...........x.............xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx
//
4.2 搜索和提取Template
其中,根据template_searcher的类型不同,搜索方式也不同:
- 在multimer中,使用hmmsearch.Hmmsearch,输入格式是sto,
pdb_seqres是PDB的数据集的序列结果。 - 在monomer中,使用hhsearch.HHSearch,输入格式是a3m,搜索
pdb70_database_path,即PDB70。 - 搜索来源不同,搜索方式不同。
源码如下:run_alphafold.py
if run_multimer_system:template_searcher = hmmsearch.Hmmsearch(binary_path=FLAGS.hmmsearch_binary_path, hmmbuild_binary_path=FLAGS.hmmbuild_binary_path, atabase_path=FLAGS.pdb_seqres_database_path)template_featurizer = templates.HmmsearchHitFeaturizer(mmcif_dir=FLAGS.template_mmcif_dir, ...)
else:template_searcher = hhsearch.HHSearch(binary_path=FLAGS.hhsearch_binary_path, databases=[FLAGS.pdb70_database_path])template_featurizer = templates.HhsearchHitFeaturizer(mmcif_dir=FLAGS.template_mmcif_dir, ...)
具体而言,pdb_seqres是PDB的数据集,参考AlphaFold-Multimer - Protein complex prediction with AlphaFold-Multimer。

搜索之后,使用template_featurizer,提取特征,搜索库都是mmcif,即199,000的PDB蛋白库,格式是cif。
逻辑位于alphafold/data/templates.py#HhsearchHitFeaturizer,源码如下:
TEMPLATE_FEATURES = {'template_aatype': np.float32,'template_all_atom_masks': np.float32,'template_all_atom_positions': np.float32,'template_domain_names': object,'template_sequence': object,'template_sum_probs': np.float32,
}
...
class HhsearchHitFeaturizer(TemplateHitFeaturizer):"""A class for turning a3m hits from hhsearch to template features."""def get_templates(self,query_sequence: str,hits: Sequence[parsers.TemplateHit]) -> TemplateSearchResult:"""Computes the templates for given query sequence (more details above)."""logging.info('Searching for template for: %s', query_sequence)template_features = {}for template_feature_name in TEMPLATE_FEATURES:template_features[template_feature_name] = []...for hit in sorted(hits, key=lambda x: x.sum_probs, reverse=True):# We got all the templates we wanted, stop processing hits.if num_hits >= self._max_hits:breakresult = _process_single_hit(query_sequence=query_sequence,hit=hit,mmcif_dir=self._mmcif_dir,max_template_date=self._max_template_date,release_dates=self._release_dates,obsolete_pdbs=self._obsolete_pdbs,strict_error_check=self._strict_error_check,kalign_binary_path=self._kalign_binary_path)...return TemplateSearchResult(features=template_features, errors=errors, warnings=warnings)
核心在于alphafold/data/templates.py#_process_single_hit(),源码如下:
features, realign_warning = _extract_template_features(mmcif_object=parsing_result.mmcif_object,pdb_id=hit_pdb_code,mapping=mapping,template_sequence=template_sequence,query_sequence=query_sequence,template_chain_id=hit_chain_id,kalign_binary_path=kalign_binary_path)
if hit.sum_probs is None:features['template_sum_probs'] = [0]
else:features['template_sum_probs'] = [hit.sum_probs]
调用_extract_template_features提取Template特征:
Parses atom positions in the target structure and aligns with the query.Atoms for each residue in the template structure are indexed to coincide with their corresponding residue in the query sequence, according to the alignment mapping provided.
解析目标结构中的原子位置,并与查询对齐。模板结构中的每个残基的原子都按照所提供的对齐映射,与查询序列中的相应残基一一对应。
输出特征:
return ({'template_all_atom_positions': np.array(templates_all_atom_positions),'template_all_atom_masks': np.array(templates_all_atom_masks),'template_sequence': output_templates_sequence.encode(),'template_aatype': np.array(templates_aatype),'template_domain_names': f'{pdb_id.lower()}_{chain_id}'.encode(),},warning)
在Template特征中,以序列长度117、Template数量20为例,包括6类特征,具体输出如下:
template_all_atom_positions,模板全部原子坐标,[N_tmpl, N_res, 37, 3],初始值是0,有坐标就设置为坐标值。
全部原子类型是37个:
atom_types = ['N', 'CA', 'C', 'CB', 'O', 'CG', 'CG1', 'CG2', 'OG', 'OG1', 'SG', 'CD','CD1', 'CD2', 'ND1', 'ND2', 'OD1', 'OD2', 'SD', 'CE', 'CE1', 'CE2', 'CE3','NE', 'NE1', 'NE2', 'OE1', 'OE2', 'CH2', 'NH1', 'NH2', 'OH', 'CZ', 'CZ2','CZ3', 'NZ', 'OXT'
] # := 37.
来源于:
all_atom_positions, all_atom_mask = _get_atom_positions(mmcif_object, chain_id, max_ca_ca_distance=150.0)pos[residue_constants.atom_order[atom_name]] = [x, y, z]
mask[residue_constants.atom_order[atom_name]] = 1.0
-
templates_all_atom_masks,模版全部原子Mask,[N_tmpl, N_res, 37],初始值是0,有坐标就设置为1。 -
template_sequence,模版序列,[N_tmpl],例如:
output_templates_sequence = ''.join(output_templates_sequence)'template_sequence': output_templates_sequence.encode(),# output
array([b'QLEDSEVEAVAKGLEEMYANGVTEDNFKNYVKNNFAQQEISSVEEELNVNISDSCVANKIKDEFFAMISISAIVKAAQKKAWKELAVTVLRFAKANGLKTNAIIVAGQLALWAVQCG',...dtype=object)
template_aatype,模版氨基酸类型,[N_tmpl, N_res, 22],22表示AA + Unknown + Gap,包含全部大写字母以及"-",数据如下:alphafold/common/residue_constants.py
HHBLITS_AA_TO_ID = {'A': 0,'B': 2,'C': 1,'D': 2,'E': 3,
...'-': 21,
}
template_domain_names,模版域名,[N_tmpl],PDB+链名,例如5jwf_A,5jwg_B。
'template_domain_names': f'{pdb_id.lower()}_{chain_id}'.encode()
template_sum_probs,模版相似性,[N_tmpl, 1],值越大,越相似,从搜索结果中,解析出来的。
源码:
# Parse the summary line.pattern = ('Probab=(.*)[\t ]*E-value=(.*)[\t ]*Score=(.*)[\t ]*Aligned_cols=(.*)[\t'' ]*Identities=(.*)%[\t ]*Similarity=(.*)[\t ]*Sum_probs=(.*)[\t '']*Template_Neff=(.*)')match = re.match(pattern, detailed_lines[2])if match is None:raise RuntimeError('Could not parse section: %s. Expected this: \n%s to contain summary.' %(detailed_lines, detailed_lines[2]))(_, _, _, aligned_cols, _, _, sum_probs, _) = [float(x) for x in match.groups()]
示例Case:
No 6883
>2G9Z_A thiamine pyrophosphokinase (E.C.2.7.6.2); Thiamin-PNP, TPK, Thiamin pyrophosphokinase, structural; HET: VNP, PO4; 1.96A {Candida albicans}
Probab=0.43 E-value=1.1e+04 Score=15.10 Aligned_cols=38 Identities=13% Similarity=-0.010 Sum_probs=22.3 Template_Neff=6.500Q seq 77 QTHLLWVPGGAPDVLRKLMRGGPYLDFLKAQSAGADHVSSVCEGALLLAA 126 (214)
Q Consensus 77 ~~d~livpgg~~~~~~~~~~~~~~~~~l~~~~~~~~~v~~i~~g~~~La~ 126 (214)...+|||.+|... . ++....+....+++...|+..|.+
T Consensus 62 ~~~~lIilng~~~--~----------~~~~l~~~~~~vI~ADGGan~L~~ 99 (348)
T 2G9Z_A 62 NHNVLLILNQKIT--I----------DLISLWKKCEIIVCADGGANSLYE 99 (348)
T ss_dssp -CEEEEECSSCCC--S----------CHHHHHTTCSEEEEETTHHHHHHH
T ss_pred CCEEEEEeCCCCC--H----------HHHHHHhcCCEEEEeCHHHHHHHH
Confidence 4566677766653 2 122233456778888888877765
最后,将初始化完成的3块特征,合计6+4+6=14类特征,合并返回即可。
def process(self, input_fasta_path: str, msa_output_dir: str) -> FeatureDict:return {**sequence_features, **msa_features, **templates_result.features}
这篇关于PSP - AlphaFold2 的 2.3.2 版本源码解析 (1)的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!






