本文主要是介绍New Journal of Physics:不同机器学习力场特征的准确性测试,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
文章信息
作者:Ting Han1, Jie Li1, Liping Liu2, Fengyu Li1, * and Lin-Wang Wang2, *
通信单位:内蒙古大学物理科学与技术学院、中国科学院半导体研究所
DOI:10.1088/1367-2630/acf2bb

研究背景
近年来,基于DFT数据的机器学习力场(machine learning force field, MLFF)发展迅速。而MLFF预测能量和力的准确性依赖于对化学环境的准确描述(特征)。目前已经提出了诸多特征方法,但是对于给定的物理问题如何选择合适的特征方法仍然是个难题。汪林望教授课题组和内蒙古大学李锋钰老师课题组使用相同的系统、相同的数据集、相同的数量特征,系统地比较了9种不同特征方法的准确性,为如何选择特征方法提供了宝贵思路。
文章简介
本文基于S、C体系,测试了9种特征类型,包括余弦特征(2-body+3-body)、高斯特征(2-body+3-body)、Moment Tensor Potential(MTP)特征、spectral neighbor analysis potential(SNAP)特征、Chebyshev多项式(DP-Chebyshev)特征和高斯多项式(DP-Gaussian)特征和原子簇展开(Atomic Cluster Expansion,ACE)特征。通过比较各特征类型对系统总能、群能、力的均方误差(RMSE)和损失函数误差,筛选出误差最小的最优特征方法为MTP,并在S、C和Cu系统中测试验证了MTP模拟的精度。
此外考虑到余弦(2-body+3-body)特征和高斯(2-body+3-body)特征对群能、总能和力模拟的较小误差,本文提出对于一个给定的系统,可以先用线性模型对不同的特征进行测试,然后将该特征集用于神经网络模型的训练。为了扩展数据集进行训练,我们采用了能量分解方法,将DFT总能分解到单个原子或者多个原子上。
01
单原子能量Ei:将DFT总能量划分为原子能量,每个原子的能量之和等于系统的总能量

由于Ei只依赖于原子i附近的原子构型,这种局部特性为MLFF模型提供了比作为总能量导数的力更加有效的数据。
02
组群多原子能量Egroup:为了减少局部能量密度分配给附近原子时的非唯一性,本文定义了一个群能(𝐸group):

为DFT计算出的围绕中心原子i的原子能量的加权平均。
主要内容
01
使用DFT进行AIMD计算产生数据集
具体步骤如表1所示。计算得到3个S训练数据集:S-300 K分子动力学轨迹,S-1500 K分子动力学轨迹,S-300 K和S-1500 K轨迹组合;以及4个不同构型的C在 300-3500K的训练数据集。相应温度下的S和C结构分别如图2和3所示。
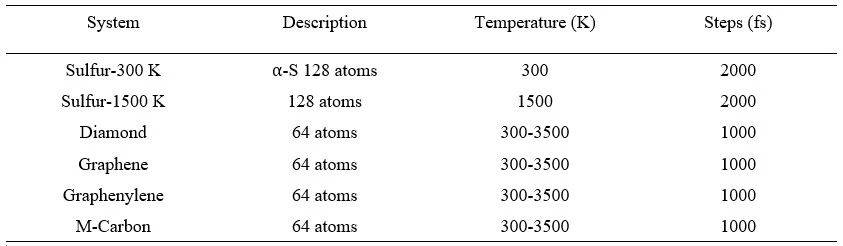
表1 S和C体系及其AIMD步骤

图2 S的结构: (a)初始s8环结构; (b) 300k下2 ps模拟后的结构; (c) 在1500 K下模拟2 ps后的结构

图3 (a)金刚石、(b)石墨烯、(c)类石墨烯 (d) m -碳结构在0 K和300-3500K下的俯视图和侧视图
02
通过拟合总能、群能、力和损失函数的误差比较不同特征方法的准确性
S-300 K和S-1500 K数据集(包含256000个原子能量和768000个原子力),S-300K和S-1500 K组合数据集(包含12000个原子能量和1536000个原子力)以及4种不同结构C-300-3500K数据集的总能、群能、力和损失函数的均方根误差分别如图4、5、6所示。
对于S-300 K体系(图4实线),MTP特征对总能的拟合效果最好(RMSE~0.060 eV);余弦(2-body+3-body)特征对群能拟合效果最好(RMSE~0.004 eV); 高斯特征(2-body+3-body)拟合力效果最好(RMSE~0.09 eV/Å)。总体而言,MTP特征为最佳线性模型,损失函数误差为0.002。
对于S-1500 K系统(图4虚线),各RMSE均大于S-300 K系统,群能、总能和力的均方根误差最小值分别在0.017 eV、0.263 eV和0.419 eV/Å左右,损失函数的拟合误差为0.036。除了力的最小误差由ACE特征得到,群能、总能和损失函数的最小误差均由MTP特征得到。
S-300 K和S-1500 K组合数据集与S-1500K数据集有类似的结果,MTP线性模型对总能量(RMSE~0.269 eV)、群能(RMSE~0.013 eV)和损失函数(RMSE~0.027)的误差最小,ACE特征是力精度的最佳特征类型(RMSE ~0.270 eV/Å)。但是由图5可知,组合数据集的各项误差均大于S-300 K的拟合结果,说明复杂的模拟系统对力场的拟合来说具有更高的挑战性。

图4 在S-300 K数据集(实线)和S-1500 K数据集(虚线)中,不同特征类型对于(a)总能、(b)群能、(c)力、(d)损失函数的训练误差。在python-ace包中使用了一个不同的无群能量损耗函数,故ACE结果仅在总能和力的图中显示
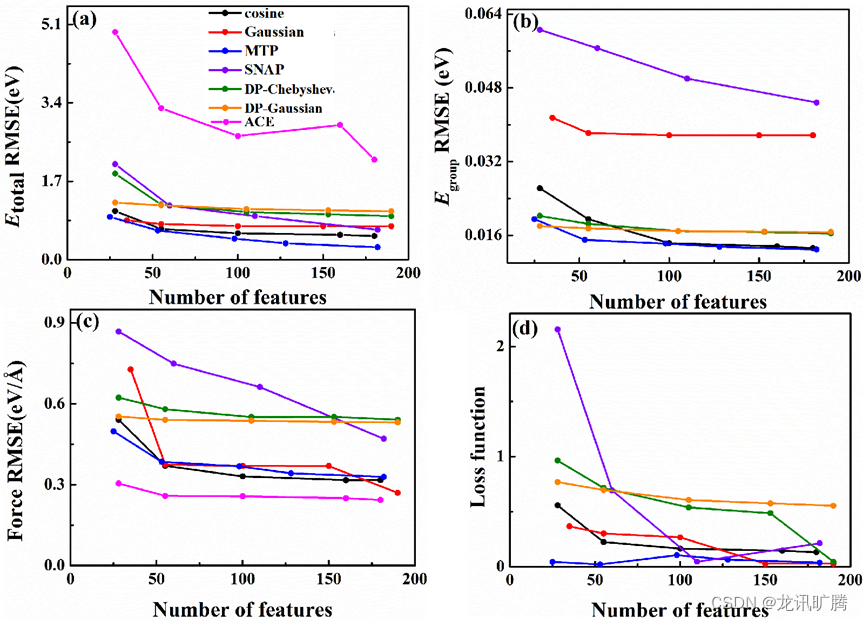
图5 在S-300 K和S-1500 K组合数据集中,不同特征类型对于 (a)总能、(b)群能、(c)力、(d)损失函数的训练误差。在python-ace包中使用了不同的无群能量损失函数,故ACE结果仅在总能量和力的图中显示
对于C系统,ACE模拟存在很大的错误,故只讨论其他6种特征方法的结果。如图6所示,MTP线性模型在拟合群能(RMSE~0.101 eV)、总能(RMSE~0.012 eV)方面表现出优势; 高斯(2-body)特征和余弦(3-body)特征在拟合力(RMSE~0.110 eV/Å)方面具有优势; MTP特征在损失函数上误差接近(RMSE~0.016)。

图6 在C系统的组合数据集中,不同特征类型对于 (a)总能,(b)群能,(c)力和(d)损失函数的训练误差
03
选择最优特征方法MTP,对测试集进行训练
对于S系统:使用不在训练集内的S结构,利用DFT分别在300K和1500K下进行6ps 的AIMD计算,并以最后1ps为测试集。用S-300K、S-1500K和S-300-1500K模型对两个测试集进行模拟。其与DFT计算相比,训练模型和测试集在同一温度下时,总能和力都有很好的匹配性,如图7和8所示。此外,当训练模型和测试集不在同一温度范围,模拟的误差非常大,这是由于不同温度下训练模型与测试集中的结构信息不同。

图7 基于相同轨迹的MLFF模型和DFT计算能量的比较:基于S-300K模型(a, b)、S-1500K模型(c, d)和组合模型(e, f)对S-300K-new集(a, c, e)和S-1500K-new集(b, d, f)

图8 基于相同轨迹的MLFF模型和DFT计算受力的比较: S-300K模型(a, b)、S-1500K模型(c, d)和组合模型(e, f)对S-300K-new (a, c, e)和S-1500K-new (b, d, f)的影响
对于C系统:选择训练集之外的C结构进行了300K 6ps的NVT AIMD模拟,以最后1ps轨迹作为测试集。如图9 (a) 和 (b) 所示,MTP-LR模型在AIMD轨迹上的总能量和力也与DFT吻合良好。C体系的总能和力的误差分别为0.22eV和0.09 eV/Å。
对除C和S以外的Cu系统:以Cu-300-1000 K的NVT 2 ps AIMD模拟作为训练集;在1000 K时对训练集之外的Cu结构进行了1 ps的NVT AIMD仿真,作为测试集。如图9 (c)和 (d) 所示,可见Cu系统的MLFF模型沿着AIMD轨迹的总能和力都与DFT结果吻合较好。其中,Cu体系的总能和力的误差分别为0.12 eV和0.05 eV/Å。
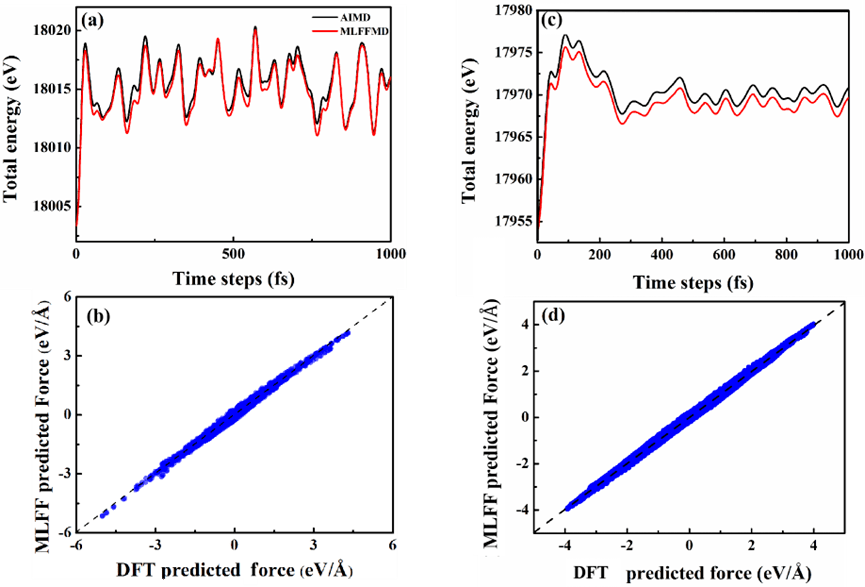
图9 在C (a, b)和Cu (c, d)两种模型中,DFT和MTP线性模型沿分子动力学轨迹的总能和力的比较
这篇关于New Journal of Physics:不同机器学习力场特征的准确性测试的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!






